
.jpg)
el concepto discapacidad se refiere a la condición de vida de una persona, que obstaculiza su funcionamiento intelectual, sensorial y motriz, afectando su desarrollo psicomotor, cognoscitivo, de lenguaje y socioafectivo. Estas limitaciones se manifiestan en dificultades para aprender, adquirir conocimientos y lograr su dominio y representación; por ejemplo: la adquisición de la lectura y la escritura, la noción de número, los conceptos de espacio y tiempo, las operaciones de sumar, restar, multiplicar y dividir.
Características del niño
con discapacidad intelectual
La discapacidad intelectual puede presentarse en el ser humano antes del nacimiento, duranteel parto o durante los cinco primeros años de vida, como resultado de altas temperaturas que
producen meningitis y convulsiones, es decir, contracciones violentas e involuntarias que afectan
el funcionamiento del cerebro; también por un traumatismo derivado de un golpe fuerte en el cerebro,
que ocasiona diferentes formas y características de la discapacidad intelectual. Las discapacidades
de tipo intelectual más comunes son:
Trastornos cromosómicos
• Síndrome de Down.

Se trata de una alteración genética ocasionada por la presencia de un cromosoma extra en el par 21, y se produce durante la división celular en el momento de la gestación, sin que alguno de los padres sea responsable de que esto suceda. Los niños con síndrome de Down presentan rasgos físicos similares, de modo que se parecen mucho entre sí, y enfrentan una condición de vida diferente, no una enfermedad. Tres características distinguen a los niños: bajo tono muscular, discapacidad intelectual y retardo en el lenguaje. Las alteraciones cromosómicas llevan el apellido del médico que las descubrió, en este caso el doctor John Langdon Down (en 1866); otros síndromes, los de Raid y West, aunque poco comunes, también conllevan discapacidad intelectual. Síndrome significa conjunto de características.
Tratamiento y pronóstico del síndrome de
Down
No existe tratamiento para el síndrome de
Down, salvo los programas de integración y de educación especial dirigidos al
desarrollo de las capacidades intelectuales del niño.
La supervivencia de los pacientes con
síndrome de Down depende de la gravedad de las malformaciones viscerales: estas
malformaciones determinan el fallecimiento de muchos de ellos en los primeros
años de vida, de modo que los pacientes mayores de cinco años tienen ya
expectativas de vida razonablemente largas (entre 50 y 60 años).
Los peligros secundarios que amenazan a
los niños mayores y a los adultos son el fácil desarrollo deleucemias (el
riesgo está aumentado 20 veces respecto a lo normal) y el desarrollo de unaenfermedad
de Alzheimer muy precoz.
¿Qué es el síndrome del cromosoma X frágil?
El síndrome del cromosoma X
frágil (FXS) es la causa hereditaria,es decir que se pasa de padres a hijos, más frecuente de discapacidad intelectual
(conocida previamente como retraso mental). Se estima que el
síndrome del cromosoma X frágil afecta a aproximadamente 1 de 4,000
niños y 1 de 6,000 a 8,000 niñas. Tanto los niños como las
niñas pueden presentar este síndrome pero por lo general
afecta un poco más a las niñas.
¿Qué causa el síndrome del cromosoma X frágil?
La causa del síndrome del
cromosoma X frágil es genética. Ocurre cuando hay un cambio en un gen en
el cromosoma X llamado FMR1. El gen FMR1 produce una
proteína necesaria para el desarrollo normal del cerebro.
Con el síndrome del cromosoma X frágil, el gen FMR1 no funciona
de manera adecuada y al no producir esta proteína, el
cerebro no funciona como debería. La falta de esta proteína causa el
síndrome del cromosoma X frágil.
¿Qué afecciones son más comunes
entre los niños con síndrome del cromosoma
X frágil?
Los niños con el síndrome del
cromosoma X frágil pueden tener problemas de aprendizaje,
retrasos en el habla y el lenguaje yproblemas de comportamiento como
el trastorno de déficit de atención e hiperactividad (ADHD,
por sus siglas en inglés) y ansiedad. Algunos niños mayores pueden
exhibir conductas agresivas. También pueden presentar depresión.
Los niños con el síndrome del cromosoma X frágil por lo
general padecen de discapacidad intelectual que oscila entre leve
y grave. Muchas niñas con este síndrome tienen una inteligencia normal.
Otras tienen un cierto grado de discapacidad intelectual con o
sin problemas de aprendizaje. Los trastornos del espectro
autista (TEA) ocurren con más frecuencia en niños que tienen el síndrome
del cromosoma X frágil.
características

síndrome de Turner
El síndrome de Turner se
define como un trastorno genético causado por una alteración (por la falta total o parcial) del cromosoma X. Los seres humanos tenemos 46
cromosomas, que son pequeñas estructuras en forma de bastón que contienen la
información genética o ADN que se encuentran presentes en el núcleo de todas
las células vegetales y animales. De estos 46 cromosomas hay dos que determinan
el sexo de los individuos: el X y el Y. Las mujeres poseen dos cromosomas X,
uno heredado del padre y el otro de la madre. Por su parte, los hombres tienen
un cromosoma X heredado de la madre y un cromosoma Y heredado del padre.
Por todo ello, esta enfermedad genética
sólo afecta a las niñas, ya que en los niños, al tener sólo un cromosoma X, la
ausencia total o parcial del mismo sería incompatible con la vida.
La causa
exacta
por la cual se produce este trastorno cromosómico no se conoce bien, aunque se
apuntan dos posibilidades. Por un lado, podría deberse a un error en la
división de las células sexuales (meiosis), ocurrido en el momento de
formarse el óvulo o los espermatozoides, que haga que uno de los dos no porte
el cromosoma X. Por otro lado, también se baraja la opción de que la pérdida
del cromosoma se produzca más adelante, en la división del óvulo ya fecundado (mitosis),
inmediatamente después de la concepción. La frecuencia con la que este síndrome
se presenta en la población es de 1 entre 2.500 recién nacidos vivos del sexo
femenino.
El Síndrome de Turner debe su nombre al
Dr. Henry Turner, médico que lo describió por vez primera en el año 1938. Este
trastorno también es conocido como Síndrome 45, X; Síndrome Bonnevie-Ulrich;
Síndrome Morgagni-Turner-Albright o Monosomía X, entre otros tantos.
Tratamiento del síndrome de Turner
Los pacientes con Síndrome de Turner
deben ser evaluados y tratados periódicamente por un grupo multidisciplinario
conformado por varios especialistas: pediatra, cirujano, nefrólogo, cardiólogo,
psicólogo, nutricionista… Los aspectos más relevantes se tratan de la siguiente
forma:
- La talla baja, uno de los principales problemas clínicos del Síndrome de Turner, se trata con hormona de crecimiento (a pesar de que las pacientes producen esta hormona). Esta hormona se ha desarrollado por ingeniería genética y es igual a la hormona de crecimiento que sintetizamos en nuestro cuerpo. Se suele recomendar la aplicación diaria de una inyección subcutánea, se debe iniciar estas inyecciones a partir de los cuatro años de vida. Si no se administra esta hormona la talla final de las pacientes con Síndrome de Turner puede ser hasta 20 centímetros menor que la del resto de la población femenina, pero si la hormona de crecimiento se administra apropiadamente esta diferencia se puede reducir a solo cinco o seis centímetros menos. Existen otros tratamientos que se pueden aplicar junto con la hormona de crecimiento que consiste en el alargamiento quirúrgico de las extremidades o alargamientos óseos. La evolución natural de la talla en las pacientes con Síndrome de Turner tiene un patrón típico:
- Fase de crecimiento prenatal (dentro del útero materno): ocurre retraso del crecimiento por lo que al nacer tienen una talla que es de dos a tres centímetros menor que la de las demás recién nacidos del sexo femenino.
- Etapa recién nacido y primer año de vida: durante esta etapa sigue retrasándose su crecimiento en relación a la talla por lo que nunca alcanzan los percentiles en las tablas de crecimiento de desarrollo para su edad y sexo. Al cumplir el primer año tienen unos 10 centímetros menos que el resto de las niñas.
- Etapa preescolar y escolar: mantienen la situación del retraso de crecimiento.
- Etapa preadolescencia y adolescencia: al llegar a los 12 años aproximadamente, la talla de estas niñas se encuentra por debajo de la media y durante la etapa del desarrollo no se presenta el típico estirón, ya que no producen las hormonas que se necesitan para que esto ocurra.
- Etapa adulto joven: a pesar de estar con una talla más baja que el resto de las chicas, el proceso de crecimiento se mantiene por más tiempo activo y esto sucede por un retraso en el cierre de los puntos de osificación, esto se traduce en que las pacientes con Síndrome de Turner pueden continuar ganado centímetros hasta los 19 a 20 años aproximadamente.
- En relación con los problemas del desarrollo se debe administrar tratamiento con estrógenos entre los 13 y 14 años para que ocurra la menarquia (aparición de la menstruación), para el desarrollo de los caracteres sexuales secundarios, para disminuir el riesgo de osteoporosis y disminuir el riesgo de enfermedades vasculares. Los estrógenos se pueden administrar a través de píldoras, implantes intradérmicos, parches en piel… Cuando ocurre la menstruación (aproximadamente dos años más tarde) se suman progestágenos al tratamiento, haciéndose éste cíclico. A pesar de esta terapia, las mujeres con Síndrome de Turner no suelen embarazarse de forma espontánea, con la consecuenteinfertilidad, sin embargo pueden tener hijos a través de las técnicas de fertilización in vitro(trasferencia de embriones) con la misma facilidad que otra mujer.
- La talla baja y el fallo en los ovarios son factores de riesgo de osteoporosis, por ello hay que asegurar un buen aporte de calcio y vitamina D.
- Si la paciente presenta muchas alteraciones en cara y cuello se debe analizar la posibilidad de la cirugía plástica para atenuar estos rasgos.
- Debe vigilarse el peso y la dieta para disminuir el riesgo de obesidad y diabetesasociada, que si llega a presentarse puede tratarse con hipoglucemiantes orales.
Síndrome de Klinefelter
También conocido como Síndrome
47-XXY, el síndrome de Klinefelter se define como un trastorno
cromosómico que afecta el desarrollo sexual masculino. En
1942, el Dr. Harry Klinefelter y sus compañeros de trabajo en el Hospital
General de Massachussets, en la ciudad de Boston, publicaron un informe
especial sobre nueve hombres que tenían aumento del tamaño de las mamas, vello
facial y corporal escaso, testículos pequeños, e incapacidad para producir
esperma.
¿Cuál es la causa del síndrome de Klinefelter?
En los años setenta, los investigadores de todo el mundo
trataron de identificar la causa de este síndrome y realizaron estudios de
cariotipo (mapa de los cromosomas) a más de 40.000 recién nacidos masculinos.
Encontraron que la prevalencia de este síndrome es de un caso por cada 500 a
1.000 recién nacidos vivos varones y que los niños con el síndrome de
Klinefelter tenían una copia extra del cromosoma X en cada célula, es decir su
cariotipo era 47, XXY.
Existen también variantes del síndrome de
Klinefelter que incluyen más de un cromosoma X extra o copias extras de ambos
cromosomas X Y en cada célula, como ocurre en el llamadocaso mosaico. Estos
pacientes suelen tener signos y síntomas más severos que el clásico síndrome de
Klinefelter, además de afectar el desarrollo sexual masculino tienen problemas
con el aprendizaje, características faciales distintivas, anormalidades
esqueléticas, problemas de coordinación de movimientos, y severos problemas
del lenguaje.
Síntomas del síndrome de Klinefelter
Estos son los signos más comunes que
presentan los pacientes afectados por el síndrome de Klinefelter:
- Los hombres con síndrome de Klinefelter tienen típicamente testículos muy pequeños que no son capaces de producir testosterona, la cual es la hormona que dirige el desarrollo sexual masculino antes del nacimiento y durante la pubertad.
- La disminución de las cantidades de testosterona durante la pubertad puede producir un aumento del tamaño de las glándulas mamarias (ginecomastia), disminución del vello corporal, de la barba y, por último, incapacidad para tener hijos, ya que no producen espermatozoides (azoospermia e infertilidad).
- Disminución del deseo sexual (libido).
- Presentan además sobrepeso, ya que tienen tendencia a la acumulación de grasa sobre todo a nivel de las caderas.
- Niños y jóvenes que padecen este síndrome suelen ser de talla alta con respecto a los de su misma edad.
- Los afectados con el síndrome de Klinefelter pueden también tener problemas de aprendizaje y dificultades con el desarrollo del lenguaje. Asimismo, tienden a ser tímidos, callados, sensibles y con escasa capacidad de establecer juicios.
Pronóstico y tratamiento del síndrome de Klinefelter
Debido a que el síndrome de Klinefelter
es un problema cromosómico, no existe un tratamiento para la enfermedad,
aunque sí puede tratarse la mayoría de los síntomas.
En general el pronóstico del
síndrome de Klinefelter es bueno, siempre y cuando el paciente esté bajo
estricta monitorización médica, que permita el tratamiento precoz de cualquier
problema que ocurra. Muchos hombres con afectados con este síndrome logran
llevar una vida plena activa y normal.
Sin embargo, los pacientes con síndrome
de Klinefelter tienen un mayor riesgo de padecer ciertas enfermedades,
tales como: diabetes tipo 1, lupus, hipotiroidismo, cáncer
de mama masculino, linfoma no-Hodgkin, obesidad y osteoporosis.
Por este motivo el médico puede programar ciertas pruebas diagnósticas como la
glucemia, las hormonas tiroideas, ecografías mamarias, etcétera, para detectar
a tiempo los problemas mencionados.
A causa de estas enfermedades y sus
posibles complicaciones, una persona con síndrome de
Klinefelter puede
tener mayor riesgo de muerte precoz.
Uso de la testosterona
Lo ideal es que los varones con síndrome
de Klinefelter comiencen el tratamiento con testosterona tan pronto como entren
en la pubertad, aunque aquellos diagnosticados en la edad adulta también pueden
beneficiarse del tratamiento con esta hormona.
Un esquema de aplicación regular de
inyecciones de testosterona producirá: aumento de la fuerza y del tamaño
muscular, y crecimiento de vello facial y corporal.
Además de estos cambios físicos, las inyecciones
de testosterona suelen provocar cambios psicológicos. A medida que comienzan a
desarrollar una apariencia más masculina, los varones con síndrome de
Klinefelter a menudo aumentan su autoestima. Muchos se vuelven más
dinámicos y enérgicos, mejoran el humor y en general el estado de ánimo. Algo
muy importante, ya que, como grupo, los niños 47, XXY tienden
a sufrir de depresión, Trastornos sindrómicos
Distrofia muscular de Duchenne
La distrofia
muscular de Duchenne (DMD) es una enfermedad genética causada por la escasez de
una proteína denominada distrofina, que es la responsable del buen
funcionamiento de la contracción muscular.
Suele aparecer en
niños de entre dos y seis años, y provoca que los músculos se debiliten hasta
el punto de que, con el tiempo, el enfermo pierde la capacidad para caminar y,
posteriormente, tiene problemas respiratorios y cardiacos.
La enfermedad de
Duchenne es una distrofinopatía que afecta a entre 20 y 30 niños de cada
100.000 varones nacidos, ya que, aunque la mujer sea portadora del gen anómalo,
no padece la enfermedad, sino que la transmite a sus hijos varones mediante el
cromosoma X.
Causas de la
distrofia muscular de Duchenne
 Se trata de una
afección de origen genético; el gen responsable de la enfermedad es recesivo y
está ligado al cromosoma X, por eso las mujeres no suelen tener síntomas (en su
caso la anomalía genética de uno de los cromosomas X se ve compensada por el
otro cromosoma X, que es normal), pero sí lo transmiten a los hijos varones
mediante el cromosoma defectuoso. Los hijos varones de las mujeres portadoras
tienen un 50% de posibilidades de padecer la enfermedad, mientras que las hijas
tienen a su vez un 50% de posibilidades de ser portadoras.
Se trata de una
afección de origen genético; el gen responsable de la enfermedad es recesivo y
está ligado al cromosoma X, por eso las mujeres no suelen tener síntomas (en su
caso la anomalía genética de uno de los cromosomas X se ve compensada por el
otro cromosoma X, que es normal), pero sí lo transmiten a los hijos varones
mediante el cromosoma defectuoso. Los hijos varones de las mujeres portadoras
tienen un 50% de posibilidades de padecer la enfermedad, mientras que las hijas
tienen a su vez un 50% de posibilidades de ser portadoras.
Cuando se
diagnostica la enfermedad a un paciente se observa que:
·
En un tercio de los casos se recoge
una clara historia familiar de la enfermedad.
·
En un tercio se observan cambios
musculares en las madres de los pacientes afectados y se considera que la
mutación ha sido materna.
·
En un tercio no se identifica
ningún familiar con afección muscular y se considera, por tanto, una mutación
del paciente.
Síntomas de la
distrofia muscular de Duchenne
El cuadro completo incluye
debilidad muscular, alteraciones de la frecuencia cardiaca y bajo coeficiente
intelectual (que no suele variar en el desarrollo de la enfermedad).
Los primeros síntomas aparecen a los dos-cuatro años de edad,
tras un desarrollo de los movimientos que suele ser normal. La debilidad
muscular comienza a apreciarse en la pelvis y las piernas, para irse
extendiendo a otras áreas del cuerpo (brazos, cuello...).
Los niños son torpes, no saltan, se apoyan con las manos en las
rodillas para incorporarse (maniobra de Gowers) de manera que trepan sobre sí
mismos, y en algunos casos resulta evidente el engrosamiento de las pantorrillas,
que es una característica típica de estos pacientes, ya que el músculo gemelo
es reemplazado por grasa y otros tejidos, por lo que la zona aumenta de tamaño
pero, como no hay músculo, se pierde la fuerza de esta zona.
Hacia los 5-6 años suelen tener caídas
frecuentes, así como dificultad para correr y subir escaleras. La debilidad de
la musculatura que rodea la columna vertebral provoca su deformación,
produciéndose escoliosis y el aumento de la curvatura normal lumbar, lo que hace que el
vientre parezca más prominente.
La capacidad de andar se pierde aproximadamente a los 10 años de
edad, debido en gran parte a las contracturas, fracturas y deformidades, por lo
que precisan una silla de ruedas para desplazarse.
La infiltración grasa aparece también en otros músculos
distintos de los gemelos, como los de los brazos, y presentan una consistencia
gomosa; sin embargo, los músculos del cráneo y de los ojos no suelen verse
afectados.
Alrededor de los 20 años aumenta la debilidad y la incapacidad
de las extremidades superiores y de los músculos respiratorios.
La muerte se produce alrededor de los 25 años de edad por la
insuficiencia respiratoria o por enfermedades del aparato respiratorio, como
la pulmonía y, en el 10% de los pacientes, a causa de la alteración cardiaca
(aunque sólo un 10% muera por esta causa, casi todos los pacientes presentan
alteración cardiaca).
Tratamiento de la
distrofia muscular de Duchenne
Todos los métodos de diagnóstico intraútero sirven actualmente en
principio para indicar un aborto, lo cual topa
con los problemas éticos. La terapia definitiva para tratar la distrofia
muscular de Duchenne está en fase de experimentación, con dos vías principales:
·
El trasplante de células musculares
normales en el músculo afectado.
·
La terapia génica, que pretende
conseguir que los músculos produzcan la distrofina de la que carecen los
enfermos.
En los últimos
años se ha visto que el tratamiento con corticoides como la prednisona, durante
12-18 meses, permite un retraso en la progresión de la enfermedad, porque
mejora temporalmente la debilidad muscular.
Los enfermos no
deben permanecer en cama de forma prolongada, y conviene estimularlos para que
mantengan una vida lo más normal y completa posible, evitando la obesidad y
practicando ejercicio mientras sea posible. Es preciso, por tanto, facilitar al
paciente una educación adecuada y prepararlo para una ocupación sedentaria. Además,
se tratarán los síntomas y las complicaciones que vayan apareciendo, como
fracturas, contracturas, infecciones pulmonares y descompensación cardiaca.
Mientras no haya
una terapia efectiva para curar la enfermedad, los pacientes deben recibir el
apoyo necesario para mejorar en todo lo posible su calidad de vida. Es muy
importante el entrenamiento del paciente y de su familia en los ejercicios de
estiramiento, que sirven para evitar o retrasar las retracciones y
deformidades, que producen dolores y deformidades y afectan a la capacidad de
movimiento. Los aparatos ortopédicos que sostengan las rodillas y las caderas
son útiles para prevenir las caídas y mantener más tiempo la capacidad de
andar. La cirugía correctora de la escoliosis mejora la posición y también la
ventilación.
La evaluación de
la función respiratoria debe hacerse desde el diagnóstico cada 6-12 meses. Las
alteraciones respiratorias, que se dan principalmente durante el sueño, mejoran
mucho con la ventilación asistida, que prolonga la supervivencia de los
pacientes.
A este respecto,
los enfermos y sus familias deben ser informados sobre la indicación y la
retirada de la ventilación asistida, para lo cual los documentos de voluntades
anticipadas son muy convenientes.

¿Cómo son los adultos
que tienen SPW?
Neurólogo
¿Por qué? Para poder realizar una valoración
y un diagnóstico neurológico que
permita orientar la intervención en la estimulación
precoz, fisioterapia, aprendizaje
y los problemas del sueño.
Dermatólogo
¿Por qué? Algunas personas con SPW
tienden a autolesionarse rascándose la
piel hasta provocarse heridas.
Normalmente no dejan que estas heridas
se curen por sí solas.
Traumatólogo
¿Por qué? Es necesario realizar exámenes
periódicos por la tendencia a la aparición
de escoliosis, osteoporosis (disminución
de la masa ósea) u otros problemas ortopédicos
que se dan con mayor frecuencia
en este síndrome.
Digestivo
¿Por qué? Presentan trastornos de tránsito,
úlcera gástrica y dificultad para vomitar.
Odontólogo
¿Por qué? La saliva espesa y los malos
hábitos alimenticios provocan problemas
dentales desde una edad muy temprana.
Errores congénitos del metabolismo

Trastornos embriológicos de la formación cerebral Anencefalia, hidrocefalia, espinina
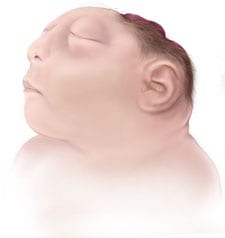 La anencefalia es un defecto de nacimiento (congénito) grave. Los bebés con anencefalia nacen con partes faltantes del encéfalo (formado por cerebro, tallo y cerebelo) y el cráneo. La anencefalia es un tipo de defecto del tubo neural (DTN). Estos defectos de nacimiento se producen durante el primer mes del embarazo, por lo general antes de que la mujer sepa que está embarazada. Al formarse y cerrarse el tubo neural, se forman el encéfalo y el cráneo del bebé (parte superior del tubo neural), la médula espinal y los huesos del espinazo (parte inferior del tubo neural).
La anencefalia es un defecto de nacimiento (congénito) grave. Los bebés con anencefalia nacen con partes faltantes del encéfalo (formado por cerebro, tallo y cerebelo) y el cráneo. La anencefalia es un tipo de defecto del tubo neural (DTN). Estos defectos de nacimiento se producen durante el primer mes del embarazo, por lo general antes de que la mujer sepa que está embarazada. Al formarse y cerrarse el tubo neural, se forman el encéfalo y el cráneo del bebé (parte superior del tubo neural), la médula espinal y los huesos del espinazo (parte inferior del tubo neural).
La anencefalia se produce cuando la parte superior del tubo neural no cierra por completo. Esto a menudo resulta en el nacimiento de un bebé sin la parte frontal del encéfalo (prosencéfalo) y la parte encargada del pensamiento y la coordinación (cerebro). Las otras partes del encéfalo a menudo no están cubiertas por hueso o piel.
Lamentablemente, casi todos los bebés que nacen con anencefalia mueren poco después del nacimiento. Los CDC calculan que todos los años nacen alrededor de 1 de cada 4,859 bebés con anencefalia
Causas y factores de riesgo
Los CDC, al igual que muchas familias afectadas, quieren saber las causas de los defectos de nacimiento. Las investigaciones nos dan pistas importantes sobre las cosas que pueden aumentar o disminuir el riesgo de tener un bebé con defectos de nacimiento, como la anencefalia. Estas pistas nos guían en la elaboración de directrices de salud pública sustentadas para la prevención.
Los CDC colaboran con muchos investigadores para estudiar los factores que pueden aumentar la probabilidad de tener un bebé con anencefalia. Los científicos creen que hay muchos factores que ejercen una influencia, como genes, conductas y medio ambiente. Investigadores de los CDC han reportado hallazgos importantes sobre algunos factores que afectan el riesgo de anencefalia:
- Un consumo insuficiente de ácido fólico antes del embarazo y en las etapas iniciales del mismo aumenta el riesgo de tener un bebé con defectos del tubo neural, como la anencefalia.2
- El número de embarazos afectados por defectos del tubo neural (espina bífida y anencefalia) ha disminuido en 27% desde que en los Estados Unidos se comenzó la fortificación de cereales con ácido fólico.2
- Los bebés de madres hispanas tienen un mayor riesgo de anencefalia, algo que no se ha podido explicar bien3.
Los CDC continúan estudiando los defectos de nacimiento, como la anencefalia, y la forma de prevenirlos. Si usted está embarazada o lo está planeando, pregúntele a su médico cómo puede aumentar su probabilidad de tener un bebé saludable.
Diagnóstico
La anencefalia se puede diagnosticar durante el embarazo o después de que nace el bebé.
Durante el embarazo
Durante el embarazo se realizan pruebas de detección (exámenes prenatales) para buscar defectos de nacimiento y otras afecciones. La anencefalia puede arrojar un resultado anormal en un análisis de suero o de sangre o puede observarse en una ecografía (la cual crea imágenes del cuerpo).
Después de que nace el bebé
En algunos casos, la anencefalia puede que no se diagnostique sino hasta después del nacimiento. La anencefalia se observa inmediatamente cuando nace el bebé.
Tratamientos
No existe una cura ni un tratamiento convencional para la anencefalia. Lamentablemente, casi todos los bebés que nacen con anencefalia mueren poco después del nacimiento.

La hidrocefalia es la acumulación de líquido en las cavidades (ventrículos) de profundidad dentro del cerebro. El exceso de líquido aumenta el tamaño de los ventrículos y ejerce presión sobre el cerebro.
El líquido cefalorraquídeo normalmente fluye a través de los ventrículos y baña el cerebro y lamédula espinal. Pero la presión de demasiado líquido cefalorraquídeo, asociado con la hidrocefalia, puede dañar los tejidos del cerebro y causar alteraciones de la función cerebral.
Aunque la hidrocefalia puede ocurrir a cualquier edad, es más común entre los niños y las personas mayores.
El tratamiento quirúrgico de la hidrocefalia puede restaurarse y mantener normales los niveles de líquido cefalorraquídeo en el cerebro. A menudo se requieren una variedad de intervenciones para controlar los síntomas o las alteraciones funcionales derivadas de la hidrocefalia.
Síntomas
Los signos y síntomas de la hidrocefalia varían generalmente según la edad de aparición. Leer más acerca de los síntomas de la hidrocefalia.
Causas
La hidrocefalia es causada por un desequilibrio entre la cantidad de líquido cefalorraquídeo se produce y cuánto se absorbe en el torrente sanguíneo. Leer más acerca de las causas de la hidrocefalia.
Factores de riesgo
En muchos casos, el evento exacto que conduce a la hidrocefalia es desconocida. Leer más acerca de los factores de riesgo de la hidrocefalia.
Complicaciones
Las complicaciones a largo plazo de la hidrocefalia pueden variar ampliamente y a menudo son difíciles de predecir. Leer más acerca de las complicaciones de la hidrocefalia.
Diagnóstico
Un diagnóstico de la hidrocefalia se basa generalmente en un examen físico, una examen neurológico y pruebas de imagen cerebral. Leer más acerca del diagnóstico de la hidrocefalia.
Tratamiento
Para tratar la hidrocefalia se puede usar la derivación o la ventriculostomía. Estas cirugías pueden presentar algunas complicaciones. Leer más acerca del tratamiento de la hidrocefalia.
Espina bífida

¿Qué es?
La espina bífida es un problema en la columna vertebral, y en algunos
casos, en la médula espinal (tejido nervioso que transmite mensajes del cerebro
a las diferentes partes del cuerpo) que presentan algunos bebés. Este problema
es uno de los más comunes que afectan a los recién nacidos.
La malformación se produce alrededor del día 28º de gestación, momento en
el que se termina de cerrar el tubo neural (parte del embrión a partir del cual
se forman el cerebro y la médula espinal).
¿Qué tipos existen?
Existen tres clases de espina bífida:

- La más grave es la que
presenta una bolsita que contiene líquido y los nervios de la médula espinal
que se asoma por el final de la columna -que se encuentra abierta. En algunos
casos la bolsita no está y la médula espinal y los nervios se pueden ver. Esto
produce parálisis en las piernas de los bebés y problemas para controlar la
vejiga y los intestinos. A este tipo de espina bífida se la conoce como
mielomeningocele.
- La menos frecuente es cuando la bolsita que se asoma al final de la
columna contiene las membranas pero no los nervios espinales. En este caso se
puede extirpar mediante cirugía permitiendo un desarrollo normal del niño. Se
la conoce como meningocele.
- La más leve se la conoce como espina bífida oculta y no produce síntomas.
Es muy común enterarse casualmente cuando el niño ya ha crecido, a través
de una radiografía por ejemplo. Básicamente consiste en la existencia de
pequeñas aberturas en algunas vértebras de la columna. Por lo general, no
requiere de ningún tratamiento.
¿Cuáles son las causas?
Se estima que una de las causas puede ser un nivel insuficiente en el
organismo de la madre de ácido fólico (una vitamina que contienen algunos
alimentos como las verduras de hojas verdes, las legumbres y las naranjas).
Otras causas pueden ser: mujeres con diabetes mal controlada o que hayan
ingerido medicamentos anticonvulsivantes durante el embarazo.
Si bien esta malformación se suele presentar en familias que no tienen
antecedentes, si éstos existen es recomendable que la pareja consulte a un
médico genetista sobre los posibles riesgos antes de concebir un futuro bebé.
En el caso de papás que ya han tenido un bebé con espina bífida -u otro
defecto del tubo neural- existe un riesgo mayor de tener otro bebé con el mismo
problema.
¿Qué problemas pueden tener los bebés y niños con espina
bífida?
Hidrocefalia:
Es la acumulación de líquido en el cerebro. Este líquido hace que el
cerebro se agrande provocando una presión que – de no ser tratada- puede
ocasionar lesiones y retraso mental. Por lo general mediante una cirugía se
coloca un tubo que permite drenarlo.
Malformación de Chiari tipo II:
Se presenta cuando el cerebro se encuentra ubicado más abajo de lo normal.
Esto provoca dificultades para respirar, tragar y debilidad en la parte
superior del cuerpo. Puede ser tratado mediante cirugías.
Médula espinal anclada:
La médula espinal normalmente se desliza con el movimiento del cuerpo hacia
arriba y hacia abajo, pero la médula espinal anclada queda retenida en su lugar
por el tejido que la rodea. Esto provoca debilidad en las piernas, curvatura de
la columna vertebral (escoliosis), dolor de espalda y piernas y alteraciones en
la vejiga. También puede ser tratada mediante cirugías.
Trastornos urinarios:
Los problemas para vaciar la vejiga por completo son frecuentes, lo que
puede provocar infecciones urinarias y lesiones en los riñones. Es importante
el tratamiento con un médico especializado en problemas urinarios.
Alergia al látex:
Debido a que es normal que estos niños tengan alergia al látex
(posiblemente por la exposición reiterada a las cirugías) se recomienda a los
médicos que utilicen en las intervenciones con estos pacientes guantes y
elementos que no sean de látex. También es aconsejable que los bebés no tengan
contacto con tetinas de mamaderas y chupetes de este material.
Problemas de aprendizaje:
Estos niños poseen una inteligencia normal generalmente, pero pueden
manifestar algunos problemas en el aprendizaje.
Otros trastornos:
Ocasionalmente pueden presentarse problemas físicos y psicológicos
adicionales, como obesidad, trastornos digestivos, depresión y problemas
sexuales.
¿Cuál es el tratamiento adecuado?
- La forma más grave (mielomeningocele) requiere cirugía dentro de las 24 a
las 48 horas del nacimiento. Esta intervención permite colocar en su lugar los
nervios que están expuestos y cubrirlos con músculo y piel. La inmediatez de
esta operación permite evitar infecciones y lesiones. Luego de la cirugía se
recomienda el tratamiento con un kinesiólogo para mejorar los problemas de
movilidad que pudieran presentar las piernas y/o los pies el bebé.
- La forma menos frecuente (meningocele) también se repara con una cirugía
y estos bebés no tienen riesgos de sufrir parálisis.
- La forma más leve (espina bífida oculta) no requiere tratamiento.
¿Cómo puede prevenirse?
Una de las principales medidas preventivas es el consumo de ácido fólico en
los meses anteriores y posteriores al embarazo lo que permite que el tubo
neural se cierre correctamente. Si bien el ácido fólico se encuentra en algunos
alimentos, es importante que el médico garantice los niveles necesarios del
mismo recetándolo como complemento vitamínico para la mujer embarazada.
-
Influencias ambientales
Desnutrición materna

La
malnutrición, en cualquiera de sus formas, presenta riesgos considerables para
la salud humana. En la actualidad, el mundo se enfrenta a una doble carga de
malnutrición que incluye la desnutrición y la alimentación excesiva,
particularmente en los países en desarrollo.
El
hambre y una nutrición inapropiada contribuyen a la muerte prematura de las
madres, lactantes y niños pequeños, y al desarrollo físico y cerebral
deficiente en los jóvenes. Al mismo tiempo, las tasas mundiales crecientes de
sobrepeso y obesidad están relacionadas con el aumento en las enfermedades
crónicas como el cáncer, las enfermedades cardiovasculares y la diabetes, todas
ellas afecciones que ponen en peligro la vida y son muy difíciles de tratar en
lugares con limitados recursos y con unos sistemas de salud que ya están
sobrecargados.
Desnutrición
·
en todo el mundo hay cerca de 115
millones de niños con insuficiencia ponderal;
·
la desnutrición contribuye a cerca de un
tercio de la mortalidad infantil;
·
el retraso del crecimiento (un indicador
de desnutrición crónica) dificulta el desarrollo de 171 millones de niños
menores de cinco años;
·
trece millones de niños han nacido con
bajo peso al nacer o prematuramente debido a la desnutrición materna u otros
factores;
·
la carencia de vitaminas y minerales
esenciales en la dieta afecta a la inmunidad y el desarrollo saludable. Más de
una tercera parte de los niños en edad preescolar del mundo presenta
deficiencia de vitamina A;
·
la desnutrición materna, un fenómeno
común en muchos países en desarrollo, lleva al desarrollo fetal deficiente y a
un mayor riesgo de complicaciones del embarazo;
·
en conjunto, la desnutrición materna y
la desnutrición del niño suponen más del 10% de la carga de morbilidad mundial.
síndrome de abstinencia alcohólica del feto

Síndrome de abstinencia neonatal
Es un grupo de problemas que ocurren en un recién nacido que estuvo
expuesto a drogas adictivas opiáceas mientras estaba en el útero de la madre.
Causas
El síndrome de abstinencia neonatal se produce porque una mujer embarazada
toma medicamentos opiáceos o narcóticos como la heroína, la codeína, la
oxicodona (Oxycontin), la metadona o la buprenorfina.
Éstas y otras drogas atraviesan la placenta que conecta al bebé con su
madre en el útero. El bebé se vuelve adicto junto con su madre.
Al nacer, el bebé todavía es dependiente de la droga. Debido a que ya no
está recibiendo la droga después del nacimiento, se pueden presentar síntomas
de abstinencia.
El consumo de alcohol y de otras drogas durante el embarazo también puede
causar problemas en el bebé.
Los bebés de madres que consumen otras drogas adictivas (nicotina,
anfetaminas, barbitúricos, cocaína, marihuana) pueden tener problemas a largo
plazo. Sin embargo, no hay evidencia clara de un síndrome de abstinencia
neonatal relacionado con estos fármacos.
Síntomas
Los síntomas del síndrome de abstinencia neonatal dependen de:
- El tipo
de droga que la madre consumió.
- La
forma como el cuerpo descompone la droga.
- La
cantidad de droga que ella estaba tomando.
- La
cantidad de tiempo durante el cual consumió la droga.
- Si el
bebé nació a término o antes (prematuro).
Los síntomas generalmente empiezan de 1 a 3 días después del nacimiento o
pueden tardar hasta una semana en aparecer. Dichos síntomas pueden abarcar:
- Coloración
en manchas de la piel (moteado)
- Diarrea
- Llanto excesivo o chillón
- Succión
excesiva
- Fiebre
- Reflejos
hiperactivos
- Aumento
del tono muscular
- Irritabilidad
- Mala
alimentación
- Respiración
rápida
- Convulsiones
- Problemas
para dormir
- Aumento
lento de peso
- Nariz
tapada, estornudo
- Sudoración
- Tiritar
(temblores)
- Vómitos
Pruebas y exámenes
Muchos otros trastornos pueden ocasionar los mismos síntomas del síndrome
de abstinencia neonatal. Para ayudar a hacer el diagnóstico, el médico hará
preguntas acerca del consumo de drogas por parte de la madre. A la madre se le
puede preguntar qué drogas tomó durante el embarazo y cuándo fue la última vez
que las tomó.
Los exámenes que se pueden hacer para diagnosticar la abstinencia en un
recién nacido abarcan:
- Sistema
de puntuación del síndrome de abstinencia neonatal que asigna puntos con
base en cada síntoma y su gravedad. El puntaje del bebé puede ayudar a
determinar el tratamiento.
- Examen
toxicológico de las primeras deposiciones (meconio).
- Examen
de orina (análisis de orina).
Tratamiento
El tratamiento depende de:
- La
droga involucrada
- La
salud general del bebé
- Si el
bebé nació a término o fue prematuro
El equipo médico vigilará al recién nacido cuidadosamente para buscar
signos de abstinencia, problemas con la alimentación y aumento de peso. Es
posible que los bebés que vomiten o que estén muy deshidratados necesiten
recibir líquidos a través de una vena (intravenosos).
Los bebés con el síndrome de abstinencia neonatal a menudo son melindrosos
y difíciles de calmar. Los consejos para tranquilizar al bebé abarcan:
- Mecerlo
suavemente
- Reducir
el ruido y las luces
- Envolver
al bebé en una manta
Algunos bebés con síntomas graves necesitan medicamentos como la metadona y
la morfina para tratar los síntomas de abstinencia.
El objetivo del tratamiento es recetarle al bebé una droga similar a la que
la madre consumió durante el embarazo y disminuir lentamente la dosis con el
tiempo. Esto le ayuda al bebé a desacostumbrarse de la droga y aliviar algunos
síntomas de la abstinencia. La lactancia también puede servir.
Los bebés que padecen esta afección con frecuencia no se alimentan bien y
crecen lentamente. Estos bebés pueden necesitar:
- Leche
maternizada rica en calorías que proporcione mayor nutrición.
- Porciones
más pequeñas suministradas con mayor frecuencia.
Expectativas
(pronóstico)
El tratamiento ayuda a aliviar los síntomas de abstinencia.
Posibles
complicaciones
El consumo de alcohol y de drogas durante el embarazo puede
llevar a muchos problemas de salud en el bebé además del SAN, los cuales pueden
abarcar:
- Defectos
congénitos
- Bajo
peso al nacer
- Nacimiento prematuro
- Perímetro cefálico pequeño
- Síndrome de muerte súbita del lactante (SMSL)
- Problemas
del desarrollo y la conducta
El síndrome de abstinencia neonatal puede durar de 1 semana a 6 meses.
Cuándo contactar a un
profesional médico
Constate que el médico o el personal de enfermería sepan respecto a todas
las drogas que usted toma durante el embarazo.
Llame al médico o al personal de enfermería si el bebé
tiene síntomas del síndrome de abstinencia neonatal.
Prevención
Aborde el tema de medicamentos, consumo de alcohol y de tabaco con su
médico. Si está consumiendo drogas, como el alcohol y el tabaco,
solicítele ayuda al médico para suspenderlas lo más pronto posible. Si
ya está embarazada, hable con el médico respecto a la mejor manera de
dejar de consumirlas y mantenerse a salvo junto con el bebé.
radiaciones durante el embarazo


Depende del tipo de rayos-X que necesites y de cuánta radiación vayas a
recibir. Cuanta más radiación, mayor es el riesgo para tu bebé. La mayoría de
los rayos-X que se usan para hacer diagnósticos (por ejemplo, las radiografías
dentales) no generan una exposición tan alta como para poner en peligro al
bebé. Sin embargo, está demostrado que las exposiciones superiores a 10 rads
(la unidad utilizada para medir la radiación absorbida) aumentan el riesgo de
retraso mental y de anormalidades en los ojos del bebé. No obstante, no es
necesario que te preocupes: es muy raro que un diagnóstico radiológico supere
los 5 rads. Por ejemplo, la cantidad de radiación que recibe tu bebé cuando te
haces una radiografía dental es 0.01 milirads. Como 1 rad equivale a 1.000 milirads,
tendrías que hacerte 100.000 radiografías dentales para recibir 1 rad. Las
dosis estimadas de otros procedimientos son 60 milirads para una radiografía de
pecho, 290 milirads para una del abdomen y 800 milirads para una tomografía
computarizada (CT, según sus siglas en inglés). Como comparación, piensa que,
durante el curso normal del embarazo, tu bebé estará expuesto a 100 milirads de
radiación natural del sol y de la tierra.
Aunque el riesgo es bajo, los expertos a menudo recomiendan posponer los rayos-X que no sean estrictamente necesarios hasta después de dar a luz. Pero es bueno recordar que si, por cualquier razón, tu médico cree que debes hacértelos, la cantidad de radiación que reciba tu bebé será, con toda probabilidad, demasiado pequeña como para generar riesgos. Cuando vayas a hacerte la radiografía comunica al personal técnico que estás embarazada para que te protejan adecuadamente.
Causas perinatales
Trastornos intrauterinos

anemia
La anemia se refiere a
la disminución de los valores de hemoglobina en la sangre por debajo de ciertos
niveles establecidos (en realidad disminuye el tamaño y el número de glóbulos
rojos, la concentración de hemoglobina en cada uno de ellos y el valor de la
hemoglobina total). Los valores normales oscilan entre 12-16 gr. de
hemoglobina en la mujer no embarazada y 11 a 14 gr. en la
embarazada. En este artículo trataremos exclusivamente la anemia de origen
nutricional por baja ingesta de hierro, anemia ferropénica nutricional.
Anemia durante el
embarazo? Esta es una condición especial debido a que representa un subgrupo
con frecuencia afectado, con criterios diagnósticos diferentes y posibles
consecuencias sobre un testigo inocente: el bebé
Los valores de
concentración de hemoglobina durante el embarazo son discretamente menores que
los de la mujer no embarazada y se consideran normales entre 11 y 14 gr pero
hablamos de anemia durante el embarazo cuando los valores son menores de 11
gr. durante los primeros (semanas 1 a 13) y los ultimos 3
meses del embarazo (semanas 26 a 40) y menores de 10.5
gr. durante el segundo trimestre (semanas 13 a 26)
El feto se comporta como
un parásito muy eficiente y siempre obtendrá el hierro necesario proveniente de
su madre de manera que durante el embarazo la madre consumirá sus depósitos de
hierro aceleradamente (fabricar nuevos glóbulos rojos para su propio uso y el
hierro transferido al feto y su placenta para la síntesis de hemoglobina y
otros sistemas que necesitan el hierro para el funcionamiento y desarrollo celular);
por esto la madre debe compensar esta nueva demanda mediante el consumo de
hierro en su dieta o a través de suplementos para hacer frente a esta nueva e
inevitable demanda.
Generalmente la dieta no
compensa esta nueva demanda así que es rutina necesaria suplementar la ingesta
de hierro mediante el uso de preparaciones de hierro durante todo el embarazo
aun en pacientes que no manifieste anemia ¿Cual es el tratamiento? La
corrección de los niveles de hierro sanguíneos y de sus depósitos en la médula
ósea es el tratamiento indicado en la anemia ferropénica
Prevención: la mejor
estrategia es prevenir la anemia mediante una dieta adecuada, esto no solo
evitaría la anemia sino todas aquellas enfermedades asociadas con la
desnutrición. Esto parece fácil pero recuerda que la mayor parte de la
población mundial vive en condiciones nutricionales limítrofes o
francamente precarias. Los gobiernos son los responsables de la nutrición de
sus pueblos.
El uso de suplementos de
hierro antes, durante y después del embarazo previene la anemia y sus
complicaciones
Tratamiento lento: Una vez
diagnosticada la presencia de anemia usualmente utilizamos preparaciones
ferrosas de administración por vía oral en dosis un poco más altas que la dosis
rutinaria preventiva. La mayor parte de los pacientes son manejadas de esta
forma si se hace el diagnostico precozmente, los valores de hemoglobina son
superiores a 8.5 gr, la paciente es asintomática o la fecha del parto no
esta muy cercana
Tratamiento rápido: en casos de anemia
severa con valores menores a 8.5 gr. y en presencia de
pacientes sintomáticas o con fechas de parto muy cercanas escogemos
tratamientos rápidos que pueden incluir el uso de hierro por vía intramuscular,
intravenosa
Tratamiento inmediato: recurrimos a transfusiones
sanguíneas en la paciente con anemia severa (casi siempre en pacientes
sintomáticas con valores menores a 6 gr. ) especialmente
si nos encontramos en la fecha del parto o la cesárea. Posteriormente la
paciente debe consumir suplementos de hierro para corregir su déficit de hierro
circulante y llenar sus depósitos para evitar la recurrencia de la anemia

El parto pretérmino
aparece en el 5-10% de las embarazadas; se define como el parto que se produce entre las semanas 28 a 37 del embarazo, y es un problema no
sólo obstétrico, sino también neonatal, ya que se asocia con un alto índice de
problemas para el recién nacido, incluyendo la muerte del mismo.
Otro caso es el parto inmaduro, que es
aquel que se da entre las semanas 20 y 28 de embarazo.
Cuanto más prematuro es el bebé, existen menos posibilidades de que sobreviva y, en caso
de conseguirlo, tendrá que hacer frente a mayores dificultades: retraso mental, parálisis cerebral, problemas respiratorios, digestivos, pérdidas de
visión y audición, retrasos en el desarrollo, y problemas de aprendizaje y de
conducta.
Factores de riesgo para un parto
prematuro
No se conoce una única
causa, pero existen factores de riesgo que predisponen al parto pretérmino, y que hay
que tratar de prevenir o evitar. Los más importantes son:
·
Enfermedades de la madre: infecciones de las vías urinarias, vaginales o sistémicas, enfermedades
renales, cardiacas, diabetes, anemias severas, alteraciones tiroideas...
·
Trabajo duro y estresante.
·
Enfermedades del embarazo (preeclampsia o diabetes gestacional), que pueden hacer aconsejable su interrupción
pretérmino.
·
Estado nutricional deficiente.
·
Nivel socioeconómico bajo.
·
Consumo de alcohol o drogas.
·
Exceso de actividad física.
En cualquier caso, el que la madre reúna
uno o más de estos factores de riesgo no significa que vaya a tener un parto
prematuro.
Síntomas del parto prematuro
·
Contracciones uterinas regulares, con o sin dolor.
·
Sensación de presión pelviana (sensación de que el niño empuja hacia abajo
o de pesadez).
·
Hemorragia vaginal.
·
Dolor de espalda o en las caderas.
·
Rotura de la bolsa de aguas.
·
Dolor de vientre (con o sin diarrea).
·
Molestias similares a las de la menstruación.
·
Cambios o aumentos de la secreción vaginal (flujo de color amarronado o
sanguinolento).
·
Cólicos abdominales (con o sin ganas de vomitar).
PRESENTACIONES ANORMALES DEL FETO

Presentación con
occipucio fetal posterior: Se trata de la anomalía obstétrica más frecuente. En
ella el feto se halla desviado en algún grado y el diámetro de la cabeza en esa
posición es mayor, lo que dificulta el paso del feto por la pelvis materna y el
parto es difícil. Así pues el obstetra debe decidir entre fórceps o cesárea
para solucionar el problema.
Presentación de cara y presentación de
frente: En la presentación de cara la cabeza está hiperextendida y la zona
adelantada es el mentón. Tanto en esta anomalía como en la presentación de
frente no es posible el parto vaginal.
Presentación de nalgas: En esta anomalía
el feto presenta las nalgas en lugar de la cabeza en su descenso pélvico hacía
el canal vaginal. El aumento de riesgo de muerte perinatal en partos con
presentación de nalgas es cuatro veces mayor al del parto cefálico.
El hecho de que se trate de partos frecuentemente
prematuros explica mejor estas tasas y la única manera de evitar complicaciones
es diagnosticar el problema lo antes posible.
En el parto de nalgas la flexión de las piernas del
feto constituye otra característica (presentación de nalgas franca o total) que
induce más o menos riesgo.
En todos ellos la manipulación obstétrica para
recolocación del feto es a veces factible pero si no lo fuera serán los fórceps
o la cesárea la solución.
PRESENTACIONES ANÓMALAS DEL FETO
Y MEDICINA BIOLÓGICA
La presentación de nalgas de un feto en el parto sugiere una pregunta
esencial que la obstetricia responde imprecisamente. ¿Cuáles son las causas
biológicas que determinan esta y las otras anomalías de la presentación fetal
en el parto?
Según la experiencia clínica oriental todas ellas forman parte de turbaciones
del embarazo asociadas a los siguientes síndromes:
1. Vacío
de Sangre y Frío en útero y bajo vientre.
Este terreno
define las alteraciones propias de los seis meses últimos del embarazo y supone
una doble debilidad sanguínea y deYang en las que éste de
presentación anómala del feto se entiende.
TRATAMIENTO
BIOLÓGICO: B-9, B-4 serán
la fitoterapia más precisa.
2. Vacío
de Qi, Vacío de Sangre, Vacío de Bazo.
Este triple síndrome define a toda mujer embarazada de débil constitución,
es decir anémica, inapetente, pálida, fatigada de pulso débil y lengua pálida.
Esta debilidad global supone una “agitación” en el feto que induce su anomalía
postural.
TRATAMIENTO BIOLÓGICO: B-4 y B-9.
3. Vacío crónico
de Sangre y Yin asociado a Vacío de Bazo.
En este terreno, idéntico al anterior pero con evidentes signos de Vacío
de Yin, es decir, de desarmonía neuroendocrina, se produce el calor
metabólico, “agitación del feto” y la anomalía postural.
TRATAMIENTO BIOLÓGICO: B-4, B-9 y B-5.
4. Finalmente
la Medicina Biológica concreta un terreno metabólico constituido por un Vacío
de Riñones e Hígado asociado aVacío de Yin y de Sangre y
asociado también a una inestabilidad de Ren y de Chong que
deriva a malnutrición del feto que termina en agitación y anomalía postural de
éste.
TRATAMIENTO BIOLÓGICO: B-4, B-9, B-5 y B-14.
Nota: En realidad todas estas llamadas
“agitaciones del feto” tienen una evidencia mayor en la amenaza de aborto, en
las hemorragias durante el embarazo y en la posibilidad de abortos prematuros
en general pero constituyen igualmente el fondo biológico en que las anomalías
de presentación fetales se producen.
alteraciones del cordón umbilical

Alteraciones en la longitud del cordón umbilical

Normalmente el cordón umbilical debe tener una longitud promedio de 50 a 55
centímetros para poder permitir el nacimiento por vía vaginal. Un cordón
umbilical extremadamente corto puede llegar a impedir que el bebé pueda
colocarse en la posición normal para el parto (cabecita hacia abajo), y también
puede ser la causa de la aparición de una hernia umbilical por la tracción que
provoca a ese nivel. Por el contrario, los cordones muy largos tienen una mayor
incidencia de enrollamiento del cordón alrededor del cuerpito o el cuello
fetal.
Gestación múltiple


Un embarazo múltiple es aquel en el que
se desarrolla más de un feto. Esto se produce como resultado de la fecundación
de dos o más óvulos, o cuando un óvulo se divide, dando lugar en este caso a gemelos,
genéticamente idénticos.
Los gemelos pueden
ser monocigóticos, en cuyo caso son
idénticos, o dicigóticos. Los monocigóticos proceden de un solo óvulo que se ha
dividido en dos partes que han seguido desarrollándose por separado hasta
formar a dos bebés genéticamente idénticos, del mismo sexo y físicamente muy
similares.
Los gemelos dicigóticos, por el contrario, son el resultado de una fecundación múltiple, es decir,
que la madre tenía más de un óvulo, y que estos óvulos fueron fecundados por
distintos espermatozoides por lo que, aunque se concibieran al mismo tiempo,
son bebés totalmente distintos, que podrían ser del mismo sexo, o no, y cuyo
parecido físico es semejante al de los hermanos nacidos en fechas diferentes.
Aunque es poco común, y resulta
peligroso, tanto para la madre como para los bebés, puede ocurrir también que
haya tres, cuatro, cinco embriones, o incluso más (esto es especialmente raro).
En este caso los bebés también pueden ser genéticamente idénticos, diferentes,
o una combinación de ambos tipos.
En las dos últimas
décadas se ha producido un aumento espectacular de embarazos múltiples a
consecuencia, sobre todo, del uso extensivo de técnicas de fertilidad. Existen diversos factores que influyen en que se
produzca un embarazo de este tipo:
·
Genética: los antecedentes
familiares de embarazo múltiple aumentan las posibilidades de que se repita.
·
Embarazo tardío: a mayor edad la
mujer tiene más probabilidad de una doble ovulación, especialmente entre los 30
y los 35 años.
·
Embarazos previos: si la mujer ha
tenido varios embarazos, la posibilidad de tener mellizos aumenta.
·
Tratamientos de fertilidad: los medicamentos
que se administran para mejorar la fertilidad, que estimulan a los ovarios para
que produzcan múltiples óvulos, y las técnicas de reproducción asistida, en las
que se transfieren al útero varios embriones (como la fecundación in vitro), incrementan considerablemente la incidencia de embarazo múltiple.
Los embarazos
múltiples se consideran de alto riesgo, tanto para la madre como para los fetos. Cuando hay más de dos embriones,
especialmente a partir de cuatro, a veces se aconseja a los padres una
reducción embrionaria, que consiste en inyectar una sustancia en uno o más
embriones para eliminarlos y facilitar así la viabilidad del resto. Lo mejor,
no obstante, es evitar un gran número de embriones para no tener que recurrir a
esta medida; por ejemplo, no implantar más de dos óvulos fecundados en el útero
cuando se utiliza el método in-vitro.
Trastornos neonatales
hemorragia intracraneal
Hemorragia
intracerebral, Hemorragia intracraneal, intracerebral o hipertensiva es un sangrado
en el cerebro causado por la ruptura de un vaso intracraneal. Ver también accidente cerebrovascular hemorrágico.
SÍNTOMAS DE LA HEMORRAGIA INTRACEREBRAL
·
Dolor de cabeza
·
puede suceder cuando la persona está acostada
·
puede despertar a la persona
·
puede aumentar con el cambio de posición
·
puede aumentar cuando el paciente se inclina, se tensiona y cuando tose
·
Náuseas, vómitos
·
Cambio en el estado de alerta (nivel de conciencia)
·
indiferente, introvertido
·
Cambios en la visión
·
cualquier cambio en la visión
·
disminución de la visión, pérdida total o parcial de la visión
·
Cambios en las sensibilidad
·
Dificultad al escribir o leer
·
Cambios en los movimientos
·
dificultad para mover cualquier parte del cuerpo
·
pérdida de habilidades motoras finas
CAUSAS DE LA HEMORRAGIA INTRACEREBRAL
El sangrado interno se puede presentar
en cualquier lugar del cerebro y la sangre se puede acumular en los tejidos o
en el espacio que existe entre el cerebro y las membranas que lo cubren. El
sangrado puede estar aislado en parte de un hemisferio cerebral (hemorragia intracerebral lobular) o tener lugar en otras estructuras
cerebrales, como el tálamo, los ganglios basales, el puente o el cerebelo (hemorragia intracerebral profunda).
La hemorragia intracerebral puede ser
ocasionada por lesión cerebral traumática o anomalías en los vasos sanguíneos (aneurisma o angioma).
Cuando no se produce por una de estas afecciones, se asocia comúnmente con
la presión sanguíneaalta (hemorragia intracerebral hipertensiva). En algunos casos no se encuentra
ninguna causa.
La sangre irrita los
tejidos cerebrales, ocasionando hinchazón (edema cerebral) y se puede acumular
en forma de una masa denominada hematoma. Tanto el edema cerebral como el
hematoma que se encuentran en el interior del cerebro aumentan la presión sobre
los tejidos cerebrales y los pueden destruir rápidamente.
Los síntomas varían
según la localización del sangrado y la cantidad de tejido cerebral afectado.
Por lo regular aparecen súbitamente, sin previo aviso y con frecuencia cuando
la persona está en actividad. Estos síntomas pueden aparecer ocasionalmente de
una manera escalonada, episódica o pueden empeorar de manera progresiva.
Epilepsia Neonatal
Las crisis epilépticas neonatales ocurren con mucho mayor frecuencia que en
cualquier otro período de la vida, teniendo una incidencia variable entre el
0,15 al 1,4% de los recién nacidos vivos, cifra que en los prematuros de 36
semanas o menos aumenta a cerca del 6%. • Rev. Ped. Elec. [en línea] 2005, Vol
2, N° 1. ISSN 0718-0918• Son la emergencia neurológica más común en este
periodo de la vida – 2 -3 / 1000 RNT – 50-130 / 1000 RNPT – 1-2 % de RN
ingresados a UCI neonatales – Valores basados en hallazgos clínicos – Crisis no
clínicas: incidencia desconocida
4. Bases neurobiológicas• Cerebro Fetal- RN es distinto: – Mayor densidad
neuronal – Mayor número de conexiones sinápticas – Fallo de Bomba Na-K –
Alteración de la permeabilidad al Na en la membrana neuronal – Mayor expresión
de receptores excitatorios (NMDA; AMPA) – Mayor nivel excitatorio (+ sinapsis)
glutamato y aspartato – Receptores GABA, en este periodo, serían excitatorios.
• New concepts in neonatal seizures. Neuroreport 2002; 13: A3-A8
5. • Entonces, las Crisis Convulsivas Neonatales: – Inhiben crecimiento
cerebral – Modifican circuitos neuronales – Aumentan excitabilidad neuronal –
Alteración aprendizaje y memoria visuoespacial• Tienen influencia en el
resultado del neurodesarrollo: • Predisponiendo a complicaciones tardías •
Cognitivos • Conductuales • Epilepsia
Trastornos respiratorios
Meningitis
Es una infección bacteriana de las membranas que cubren el cerebro y la
médula espinal (meninges).
Causas
Las causas más comunes de meningitis son las infecciones virales que
generalmente mejoran sin tratamiento. Sin embargo, las infecciones meningíticas
bacterianas son extremadamente graves y pueden producir la muerte o daño
cerebral incluso con tratamiento.
La meningitis también puede ser causada por:
- Irritación
química
- Alergias
a medicamentos
- Hongos
- Parásitos
- Tumores
La mayoría de las meningitis virales se debe a enterovirus, que son virus
que también pueden causar enfermedad intestinal.
Muchos otros tipos de virus pueden causar meningitis:
- La
meningitis viral puede ser causada por el virus del herpes, el mismo
virus que puede causar el herpes labial y el herpes genital. Sin embargo, las personas con herpes
labial o genital no están en mayor riesgo de desarrollar meningitis
herpética.
- Los
virus que pueden causar paperas y VIH pueden provocar meningitis aséptica.
- Recientemente,
el virus del Nilo Occidental, que se disemina por medio de las picaduras de
mosquitos, se ha convertido en una causa de meningitis viral en la mayor
parte de los Estados Unidos.
Síntomas
La meningitis viral ocurre con más frecuencia que la meningitis bacteriana
y es más leve. Por lo general, se presenta a finales del verano y principios
del otoño. Afecta con mayor frecuencia a los niños y a los adultos menores de
30 años.
Las meningitis bacteriana es una emergencia y se necesitará tratamiento
inmediato en un hospital. Los síntomas por lo general aparecen rápidamente y
pueden abarcar:
- Fiebre
y escalofríos
- Cambios en el estado mental
- Náuseas
y vómitos
- Sensibilidad
a la luz (fotofobia)
- Dolor
de cabeza intenso
- Cuello
rígido (meningismo)
Otros síntomas que pueden ocurrir con esta enfermedad:
- Agitación
- Fontanelas abultadas en los bebés
- Disminución del estado de conciencia
- Alimentación
deficiente o irritabilidad en niños
- Respiración
rápida
- Postura
inusual con la cabeza y el cuello arqueados hacia atrás (opistótonos)
La meningitis es una causa importante de fiebre en niños y recién nacidos.
Usted no puede determinar si tienen meningitis bacteriana o viral por la
forma como se sienten; es el médico quien debe hacer esto. Busque atención
médica rápida si tiene síntomas de meningitis.
Pruebas y exámenes
El médico o el personal de enfermería lo examinarán. Esto puede mostrar:
- Frecuencia
cardíaca rápida
- Fiebre
- Cambios
en el estado mental
- Rigidez
en el cuello
Si el médico piensa que usted tiene meningitis, se debe realizar una
punción lumbar ("punción raquídea") para extraer una muestra del líquido cefalorraquídeo (conocido como
LCR) para su análisis.
Los exámenes que se pueden hacer abarcan:
Tratamiento
Se usan antibióticos para tratar la meningitis bacteriana; el tipo
específico depende de la bacteria causante de la infección. Los antibióticos no
sirven para tratar la meningitis viral.
Los antivirales se les pueden administrar a las personas con meningitis
herpética.
Otros tratamientos abarcan:
- Líquidos
intravenosos (IV)
- Medicamentos
para tratar síntomas como el edema cerebral, el shock y
las crisis epilépticas
Expectativas
(pronóstico)
El diagnóstico y tratamiento oportuno de la meningitis bacteriana es
esencial para prevenir lesiones neurológicas permanentes. Generalmente, la
meningitis viral no es una enfermedad grave y sus síntomas deben desaparecer en
cuestión de 2 semanas sin complicaciones duraderas.
Posibles
complicaciones
- Daño
cerebral
- Acumulación
de líquido entre el cráneo y el cerebro (derrame subdural)
- Hipoacusia
- Hidrocefalia
- Convulsiones
Cuándo contactar a un profesional
médico
Consiga ayuda médica urgente de inmediato si piensa que usted o su hijo
presenta síntomas de meningitis. El tratamiento oportuno es clave para un buen
pronóstico.
Prevención
Ciertas vacunas pueden ayudar a prevenir algunos tipos de meningitis:
- La
vacuna contra el Haemophilus (vacuna HiB) en los niños ayudará a
prevenir un tipo de meningitis bacteriana.
- La
vacuna antineumocócica conjugada es ahora una vacuna de rutina en la
infancia y es muy eficaz para prevenir la meningitis neumocócica.
Los miembros del hogar y otros en estrecho contacto con personas que tengan
meningitis meningocócica deben recibir antibióticos preventivos para evitar
infectarse.
La vacuna meningocócica se recomienda para:
- Adolescentes
en edades de 11 a 12 años y adolescentes que ingresan a la secundaria
(alrededor de los 15 años) y que aún no hayan recibido la vacuna. Se
aplica una vacuna de refuerzo entre las edades de 16 y 18 años.
- Todos
los estudiantes universitarios que no hayan sido vacunados y que estén
viviendo en residencias estudiantiles.
- Niños
de dos años en adelante que no tengan el bazo o que tengan otros problemas
con el sistema inmunitario.
- Personas
que viajan a países donde son comunes las enfermedades causadas por el
meningococo (preguntarle al médico).
Algunas comunidades realizan campañas de vacunación después de un brote de
meningitis meningocócica.
Encefalitis

Es la irritación e hinchazón (inflamación) del cerebro, casi siempre debido
a infecciones.
Causas, incidencia y
factores de riesgo
La encefalitis es una enfermedad poco común. Se presenta casi siempre en el
primer año de vida y disminuye con la edad. Las personas muy jóvenes y los
ancianos son más propensos a presentar un caso grave.
La causa más frecuente de la encefalitis es una infección viral y muchos
tipos de virus la pueden provocar. La exposición a los virus puede suceder a
través de:
- Inhalación
de las gotitas respiratorias de una persona infectada
- Alimentos
o bebidas contaminados
- Picaduras
de mosquitos, garrapatas y otros insectos
- Contacto
con la piel
Los diferentes virus se presentarán en diferentes lugares y muchos casos
tenderán a agruparse en una cierta temporada.
La encefalitis causada por el virus del herpes simple es la causa principal
de los casos más severos en todas las edades, incluyendo los recién nacidos.
Muchos de los virus para los cuales ahora hay una vacuna también pueden
causar encefalitis, como:
Otros virus que pueden causar encefalitis abarcan:
- Adenovirus
- Virus
de Coxsackie
- Citomegalovirus
- Virus
de la encefalitis equina oriental
- Ecovirus
- Virus del Nilo occidental
El virus causa inflamación del tejido cerebral. Este tejido se hincha
(edema cerebral), lo cual puede destruir neuronas, provocar sangrado en el
cerebro (hemorragia intracerebral) y daño cerebral.
Otras causas de encefalitis pueden abarcar:
- Una
reacción alérgica a vacunas
- Enfermedad
autoimmunitaria
- Bacterias
como la de enfermedad de Lyme, sífilis y tuberculosis
- Parásitos
como nemátodos, cisticercosis y toxoplasmosis en pacientes con SIDA y otras personas que
tengan un sistema inmunitario debilitado
- Efectos
del cáncer
Síntomas
Algunos pacientes pueden tener síntomas de un resfriado o de una infección
estomacal antes de que los síntomas de encefalitis comiencen.
Cuando un caso de encefalitis no es muy severo, los síntomas pueden ser muy
similares a los de otras enfermedades, incluyendo:
- Fiebre que no es muy alta
- Dolor
de cabeza leve
- Baja
energía e inapetencia
Otros síntomas abarcan:
- Torpeza, marcha inestable
- Confusión, desorientación
- Somnolencia
- Irritabilidad
o poco control del temperamento
- Sensibilidad a la luz
- Rigidez
del cuello y de la espalda (ocasionalmente)
- Vómitos
Los síntomas en los recién nacidos y niños pequeños pueden no ser tan
fáciles de reconocer:
- Rigidez
en el cuerpo
- Irritabilidad
y llanto con más frecuencia (estos síntomas pueden empeorar cuando se
recoge al bebé del suelo)
- Alimentación
deficiente
- La
fontanela en la parte superior de la cabeza puede sobresalir más
- Vómito
Síntomas de emergencia:
- Pérdida del conocimiento, baja reacción, estupor, coma
- Debilidad muscular o parálisis
- Crisis epiléptica
- Dolor
de cabeza intenso
- Cambio
repentino en las funciones mentales:
- estado
de ánimo "llano", ausencia de estado de ánimo ostensible o
temperamento inadecuado para la situación
- deterioro
de la capacidad de discernimiento
- inflexibilidad,
egocentrismo extremo, incapacidad para tomar decisiones o apatía hacia la
interacción social
- menor
interés en las actividades diarias
- pérdida
de la memoria (amnesia), deterioro de la memoria a corto o a largo plazo
Signos y exámenes
Un examen puede mostrar:
- Reflejos
anormales
- Aumento de la presión intracraneal
- Confusión
mental
- Úlceras
bucales
- Debilidad
muscular
- Rigidez
del cuello
- Signos
en otros órganos como el hígado y los pulmones
- Erupción
cutánea
- Problemas
del habla
Los exámenes pueden abarcar:
- Resonancia magnética del cerebro
- Tomografía computarizada de la cabeza
- Cultivo
de líquido cefalorraquídeo, hemocultivo o urocultivo (sin embargo, este
examen en pocas ocasiones es útil)
- Electroencefalograma
( EEG)
- Punción lumbar y examen de líquido cefalorraquídeo
- Exámenes
para detectar anticuerpos contra un virus (pruebas de serología)
- Exámenes
para detectar cantidades diminutas del ADN de un virus (reacción en cadena
de la polimerasa o PCR, por sus siglas en inglés)
Tratamiento
Los objetivos del tratamiento son brindarle al paciente cuidados
complementarios (reposo, nutrición, líquidos) para ayudarle al cuerpo a
combatir la infección, y aliviar los síntomas. A las personas con estados de
confusión o delirio les puede servir la reorientación y el apoyo emocional.
Los medicamentos pueden abarcar:
- Medicamentos
antivirales, como aciclovir (Zovirax) y foscarnet (Foscavir), para tratar
la encefalitis por herpes u otras infecciones virales severas (sin
embargo, en la mayoría de los casos, no hay medicamentos antivirales
específicos disponibles para combatir la infección).
- Antibióticos
si la infección es provocada por ciertas bacterias.
- Anticonvulsivos,
como la fenitoína, para prevenir crisis epilépticas (convulsiones).
- Esteroides
(como la dexametasona) con el fin de reducir el edema cerebral en algunos
casos.
- Sedantes
para tratar irritabilidad o inquietud.
- Paracetamol
(acetaminofeno) para la fiebre y el dolor de cabeza.
Si la función cerebral resulta gravemente afectada, se pueden requerir
intervenciones como la fisioterapia o la terapia del lenguaje, después de que
la enfermedad esté controlada.
Expectativas
(pronóstico)
El pronóstico varía. Algunos casos de esta enfermedad son leves, cortos y
la persona se recupera por completo; mientras que otros casos son graves y
pueden ocasionar deterioro permanente o posiblemente la muerte.
La fase aguda de la enfermedad dura normalmente de 1 a 2 semanas, con desaparición
gradual o súbita de la fiebre y de los síntomas. Algunas personas pueden tardar
varios meses para una recuperación completa.
Complicaciones
El daño cerebral permanente puede ocurrir en casos severos de encefalitis y
puede afectar:
- La audición
- La
memoria
- El
control muscular
- La
sensibilidad
- El
lenguaje
- La
visión
Situaciones que
requieren asistencia médica
Acuda a la sala de urgencias o llame al número local de emergencias (como
el 911 en los Estados Unidos) si presenta:
- Fiebre
súbita
- Otros
síntomas de encefalitis
Prevención
Los niños y los adultos deben evitar el contacto con alguien que tenga
encefalitis.
El control de los mosquitos (una picadura de mosquito puede transmitir
algunos virus) puede reducir la posibilidad de que algunas infecciones puedan
llevar a que se presente encefalitis.
- Aplique
un repelente de insectos que contenga el químico DEET cuando salga a áreas
abiertas (pero nunca use productos con DEET en bebés menores de dos
meses).
- Elimine
cualquier fuente de agua estancada (como neumáticos viejos, latas, canales
y estanques de poca profundidad).
- Use
camisas de manga larga y pantalones cuando esté afuera, particularmente al
anochecer.
Vacune los animales para prevenir la encefalitis causada por el virus de la
rabia.
Las vacunas humanas que están disponibles abarcan:
- Una
vacuna para prevenir una forma de encefalitis viral que a menudo afecta a
las personas que viven en dormitorios o en el ejército
- Herpes zóster
- Sarampión
Trauma Encefálico Al Nacer

El traumatismo cráneo encefálico (TEC)
directo o indirecto, fundamentalmente es una injuria focal o difusa del
parénquima encefálico (cerebro, tronco cerebral y cerebelo); asociada
frecuentemente a una injuria secundaria como la hipotensión arterial o la
hipoxia.
La incidencia pediátrica y la mayor
morbimortalidad es en: niños menores de 4 años ocasionados por accidentes
domiciliarios y, preferentemente, por caídas (de altura menor a la talla del
paciente) en relación a precipitaciones (de altura mayor a la talla del
paciente).
En Neonatos: El diagnóstico y las medidas
de urgencia se desarrollarán paralelamente en el transcurso del parto, cesárea
o tiempo después del nacimiento, con participación del pediatra neonatólogo.
En caso de TEC con o sin cefalohematoma o caput suceda-neum por vacum o forceps, con o sin diastasis de suturas, con o sin fractura craneal, puede asociarse a paro respiratorio o apnea o paro cardiaco, que obligará al pediatra, gineco obstetra, obstetriz o neurocirujano a realizar medidas necesarias para evitar mayor daño cerebral.
En caso de TEC con o sin cefalohematoma o caput suceda-neum por vacum o forceps, con o sin diastasis de suturas, con o sin fractura craneal, puede asociarse a paro respiratorio o apnea o paro cardiaco, que obligará al pediatra, gineco obstetra, obstetriz o neurocirujano a realizar medidas necesarias para evitar mayor daño cerebral.
· Causas pos-natales:

Lesiones que producen los traumas craneoencefálicos.
Los traumas se deben a heridas penetrantes en el cráneo o a la aceleración
o desaceleración rápida del cerebro, que lesiona los tejidos en el punto de
impacto, en el polo opuesto (contragolpe) y, también, difusamente en el
interior de los lóbulos frontales y temporales. El tejido nervioso, los vasos
sanguíneos y las meninges se desgarran y rompen, lo cual ocasiona la aparición
de interrupciones nerviosas, isquemia o hemorragia intracerebral y
extracerebral y edema cerebral.
Las fracturas craneales pueden lacerar arterias meníngeas o senos venosos
grandes, produciendo un hematoma epidural o subdural. Las fracturas, sobre todo
las localizadas en la base del cráneo, pueden asimismo producir una laceración
en las meninges, originando la salida de LCR por la nariz (rinorrea) o el oído
(otorrea), o bien la entrada de bacterias o aire en el interior de la cavidad
craneal.
Las lesiones iniciales al traumatismo quedan fuera del alcance del control
médico, las lesiones secundarias, que se inician en el momento de impacto pero
que se manifiestan después de un intervalo de tiempo más o menos prolongado,
tienen posibilidades de actuación terapéutica.
Contusión y laceraciones cerebrales: Estas constituyen lesiones más
graves. Dependiendo de su gravedad, con frecuencia se acompañan de heridas
superficiales graves y de fracturas localizadas en la base del cráneo o con
depresión de fragmentos óseos. Las lesiones más graves pueden producir un
acusado edema cerebral, que ocasiona rigidez de descorticación (brazos
flexionados y en aducción; extensión de las piernas y, a menudo, del tronco) o
rigidez de descerebración (mandíbulas apretadas, retracción del cuello, todas
las extremidades en extensión). Una herniación cerebralinterna
puede producir coma, hemiplejía, pupilas dilatadas y no reactivas (unilateral o
bilateral) e irregularidad respiratoria; en estos casos se debe proceder a un
tratamiento inmediato. El aumento de la presión intracraneal, particularmente
cuando se asocia a compresión o deformación del tronco encefálico, puede
provocar aumento de la PA, junto con un enlentecimiento del pulso y de la
respiración.
INFECCIONES
Encefalitis es una inflamación del cerebro y del cordón espinal normalmente
causadas por infección viral. Las enfermedades como la rabia, la poliomelitis,
y la herpes encefalitis son todas causadas por infecciones de viruses que
afectan el cerebro y el cordón espinal y que se transmiten en una variedad de
maneras. La Encefalitis arboviral se
refiere a enfermedades similares que se transmiten por los artrópodos,
principalmente los mosquitos. Aunque la mayoría de casos de infección de
encefalitis arboviral es asintomática o tiene sólo síntomas muy apacibles, la
enfermedad a veces puede dañar los nervios y puede causar daño duradero y
muerte. Los síntomas incluyen la fiebre súbita, dolor de cabeza, vómito,
anormal sensibilidad visual a la luz, tortícolis, dolor de espaldas, confusión,
adormecimiento, torpeza, irritabilidad, y dificultad al caminar.
Las encefalitides arbovirales se mantienen en la naturaleza en ciclos de
vida complejos que involucran un hospedero vertebrado no humano primario y un
vector artrópodo primario y qué normalmente no incluye a los humanos. Los
humanos y los animales domésticos pueden contraer la enfermedad cuando el virus
escapa el ciclo e infecta a un hospedero secundario. Esto puede pasar debido a
cambios ecológicos o demográficos, o debido a cambios en las poblaciones del
vector primario, del hospedero, o ambas. Muchos arboviruses que causan la
encefalitis tienen una variedad de hospederos vertebrados diferentes y algunos
se transmiten por más de un vector.
Existen cinco tipos principales de encefalitis arboviral en los Estados
Unidos:
- Encephalitis
de St. Louis (SLE) — Este virus causa la enfermedad
transmitida por mosquitos más común en los Estados Unidos. Como la mayoría
de los tipos de encefalitis viral, se transmite a los mosquitoes por aves.
El mosquito vector de la encefalitis de St. Louis se cría en áreas que
acumulan agua como los neumáticos desechados, piscinas contaminadas,
regueras a la orilla del camino, y recipientes como las pilas para pájaros
y macetas de plantas.
- Encefalitis
equina oriental (EEE) — Como el nombre sugiere, la encefalitis
equina oriental aflige a los caballos, pero también puede afectar a los
humanos. Las erupciones de la encefalitis equina oriental normalmente
ocurren en la parte Este de los Estados Unidos. Este virus infecta pájaros
que viven cerca de los pantanos de agua dulce
- Encefalitis
equina occidental(WEE) — Como la encefalitis equina oriental, este
virus afecta a los caballos y humanos. La mayoría de los casos de
encefalitis equina occidental ocurren en las llanuras centrales y
occidentales de los Estados Unidos. Este virus florece en pájaros que
viven cerca de campos irrigados y en áreas agrícolas.
- Encephalitis
de LaCrosse (LAC) se ha identificado en los estados del Medio-oeste
y Medio-atlántico. Durante un año promedio, se informan aproximadamente 75
casos de encefalitis de LaCrosse al CDC (Centers for Disease Control). La
mayoría de los casos de encefalitis ocurren en niños menores de 16 años de
edad. Los hospederos vertebrados primarios incluyen ardillas listadas,
ardillas de árbol y otros vertebrados pequeños que viven en los hábitats
del bosque.
- Encefalitis
del Nilo occidental (WNE) — Este virus apareció por primera vez en
los Estados Unidos en 1999. Normalmente se encuentra en Africa, en el
Medio Oriente, y en partes de Europa, Rusia, India e Indonesia. El virus
es muy similar al virus de St. Louis en que los pájaros son sus hospederos
animales principales.
¿Qué
es la meningitis?

La
meningitis es la inflamación de las membranas que recubren el cerebro y la
médula espinal, denominadas meninges.
Suele ocurrir cuando una infección en otra parte del cuerpo se extiende a
través del torrente sanguíneo y acaba afectando al líquido cefalorraquídeo (el líquido que circula por los espacios que hay
dentro y alrededor del cerebro y de la médula espinal). La meningitis se puede
contraer a cualquier edad.
Hay
varios tipos distintos de meningitis, y tanto su gravedad como su tratamiento
varían en función del tipo concreto que se padezca. La mayoría de los casos de
meningitis están provocados por virus (meningitis viral) o
por bacterias (meningitis
bacteriana), pero los hongos y otros organismos
también pueden causar meningitis infecciosas. Algunos casos de meningitis están
ocasionados por traumatismos craneoencefálicos, por ciertos cánceres u otras
enfermedades o por reacciones a determinados medicamentos.
La
meningitis viral está provocada por virus como los enterovirus, que abundan en verano y a principios de otoño. Estos
virus se pueden propagar a través de la saliva, las mucosidades o las heces
infectadas. Pero esto no significa que sea necesario besar a una persona
infectada o compartir un bocadillo con ella para contraer la infección. La
infección se puede contraer al tocar superficies que ha tocado antes una
persona infectada o al exponerse a las gotitas expelidas por una persona
infectada que tose o estornuda sin taparse la boca ni la nariz. Los enterovirus
se empiezan a multiplicar en el sistema digestivo y se pueden extender por todo
el cuerpo hasta provocar una meningitis.
La
meningitis bacteriana es el tipo de meningitis más grave de todos. Si no se
trata con rapidez, puede provocar lesiones cerebrales y en algunos casos
incluso la muerte. Las bacterias que causan meningitis bacterianas más a menudo
en los adolescentes sonStreptococcus
pneumoniae y Neisseria meningitidis. La gente puede propagar estas bacterias a través de
secreciones respiratorias, procedentes de la nariz y/o de la garganta, por
ejemplo, al toser, estornudar o besar a otra persona. Después de que una
persona infectada inicie el tratamiento antibiótico contra la meningitis,
seguirá pudiéndola contagiar por lo menos durante las siguientes 24 horas.
Hay una tipo de meningitis bacteriana que está
relacionada con la enfermedad de Lyme. Este tipo de meningitis suele ser menos
grave que otras formas de meningitis bacterianas y no es mortal.
Cuáles
son los signos y síntomas
La meningitis viral y la bacteriana pueden provocar
síntomas similares. Aunque puede ser difícil saber qué tipo de meningitis
padece una persona, por lo general, los médicos lo pueden dilucidar solicitando
la aplicación de pruebas médicas.
Algunos de los síntomas de ambos tipo de meningitis
(viral y bacteriana) son:
·
fiebre
·
rigidez de cuello
·
fuerte dolor de cabeza
·
sensibilidad a la luz (denominada
fotofobia)
·
vómitos
·
nauseas
·
somnolencia extrema
·
confusión
·
convulsiones
Si tú o alguien que conoces presentáis estos síntomas,
sobre todo si habéis estado en contacto con una persona que padecía meningitis,
aseguraros de hablar con un médico. Es muy importante tratar la infección lo
antes posible.
¿Se
puede prevenir?
Lavarte las manos a conciencia y a menudo es una forma
de protegerte de la meningitis y de otras infecciones.
Aunque
la meningitis bacteriana puede asustar bastante, las probabilidades de
contraerla son bastante bajas. De todos modos, puesto que puede ser muy grave,
ahora los médicos recomiendan vacunar a todos los adolescentes contra la
meningitismeningocócica (el tipo provocado por la bacteria Neisseria meningitidis). De hecho, muchas universidades exigen que sus
alumnos estén vacunados contra la meningitis.
También
existen vacunas para otros tipos de meningitis. Si, por ejemplo, padeces alguna
afección médica que afecta a tu sistema inmunitario, es posible que tu médico
también te recomiende vacunarte contra la meningitis provocada por la
bacteria S.
pneumoniae. De todos modos, no existen vacunas para
todos los tipos de meningitis bacterianas.
La malaria

es una enfermedad causada por parásitos que se
transmiten a través de los mosquitos. Incluso en los casos relativamente leves
puede causar fiebres altas, escalofríos, síntomas gripales y anemia grave.
Estos síntomas son especialmente peligrosos en mujeres embarazadas y niños
pequeños que padecen la enfermedad por primera vez. En los niños, la malaria
aguda puede causar retrasos mentales de por vida, y se estima que las
consecuencias económicas ascienden a miles de millones de dólares anuales por
pérdidas en materia de productividad.
Infecciones Viricas

Infecciones víricas
Un virus es un diminuto organismo infeccioso (mucho menor que un hongo o
una bacteria), que necesita de una célula viva para reproducirse. El virus se
adhiere a una célula, generalmente de un tipo específico. Una vez dentro de
ellas, libera su ADN o ARN (que contiene la información necesaria para crear
nuevas partículas de virus) y asume el control de algunos procesos metabólicos
de la misma. En consecuencia, los componentes del virus son fabricados dentro
de la célula y ensamblados adecuadamente para que el virus sea liberado y siga
manteniendo su capacidad.
Trastornos desmielinizantes (trastornos
postinfecciosos o postinmunitarios)

Síndrome del intestino irritable. Dispepsia funcional. Gastroenteritis. Infección. Inflamación. Trastorno funcional digestivo.
Los trastornos funcionales digestivos (TFD) suponen una parte importante de la patología gastroenterológica. Diversos estudios han comprobado la aparición de síndrome del intestino irritable (SII-PI) tras una gastroenteritis aguda (GEA). Se ha propuesto que infecciones no digestivas pueden aumentar el riesgo de iniciar un SII. También hay datos que demuestran que una infección digestiva puede ser el desencadenante de una dispepsia funcional (DF-PI). La posible aparición de SII-PI o DF-PI depende tanto de factores infecciosos como del huésped. Se han encontrado fenómenos de microinflamación en pacientes con TFD post-infecciosos. Los estudios realizados en modelos animales demuestran que la infección e inflamación aguda da lugar a cambios permanentes en la motilidad y la sensibilidad digestiva. La importancia de la GEA en el desarrollo de los TFD se debe no sólo a ser un excelente modelo natural para el estudio patogénico, sino que también abre la posibilidad de actuar de manera preventiva.
postinmunitarios
El sistema inmunitario es una red compleja de células, tejidos y órganos que funcionan en equipo para defendernos de los gérmenes. Ayuda a nuestros cuerpos a reconocer estos "invasores" y a mantenerlos fuera de nuestro organismo y, si no puede, encontrarlos y deshacerse de ellos.
Si nuestro sistema inmune no funciona bien, puede causar serios problemas. El resultado puede ser enfermedades entre las que se incluyen:
- Alergia y asma: respuestas inmunes a sustancias que en general no son dañinas
- Enfermedades por deficiencia inmunológica: trastornos que se producen cuando falta uno o varios de los componentes que forman el sistema inmunitario
- Enfermedades autoinmunes: trastornos que causan que el sistema inmune ataque por error a nuestras propias células y órganos.
Trastornos degenerativos
síndrome de Rett

El síndrome de Rett es una patología del desarrollo neurológico, de causa genética, que afecta principalmente a las niñas y muy rara vez a los niños. Esta enfermedad debe su nombre al médico Austriaco Andreas Rett, quien en el año 1966 describe los casos de 22 niñas que tenían movimientos repetitivos en las manos, como de “lavado de manos”, acompañado de problemas motores y retraso mental.
Muchas veces el síndrome de Rett se confunde con elautismo, la parálisis cerebral o con retrasos del desarrollo sin un origen claro.
La causa del síndrome de Rett es una alteración (mutación) en el gen MECP2 (methyl-CpG-binding protein 2), localizado en el locus (posición de un gen dentro de un cromosoma) q28 del Cromosoma X. A través de este gen se produce una proteína (llamada MeCP2) que está ampliamente distribuida a nivel del núcleo de las células y que es especialmente abundante en las neuronas maduras del sistema nervioso central. Esta proteína juega un papel importante en la regulación de la sinapsis (comunicación entre las neuronas) y es fundamental en el desarrollo del sistema nervioso tras los primeros meses de vida. Existen más de doscientas mutaciones descritas en este gen, sin embargo no se sabe con exactitud cómo actúa la proteína anormal que codifica el gen mutado para producir la enfermedad.
Aunque el síndrome de Rett es de causa genética, en cerca de un noventa y nueve por ciento de los casos la mutación aparece en el paciente espontáneamente durante su desarrollo embrionario en el vientre materno, por lo tanto no lo ha heredado de sus padres. En el uno por ciento de las familias restantes con Rett el gen defectuoso se ha transmitido desde las mujeres portadoras de la mutación hacia sus hijos. Los científicos aún están tratando de comprender el proceso de la herencia en esta complicada enfermedad.
En relación a su prevalencia, esta enfermedad se clasifica en el grupo de enfermedades rarasy se presenta en uno de cada diez mil recién nacidos vivos del sexo femenino, siendo la segunda causa más frecuente de retraso mental en este sexo.
Usualmente se diagnostica durante los dos primeros años de vida, lo que es fundamental para indicar tratamientos dirigidos a mejorar en retraso psicomotor que presentan los pacientes, ya que los cambios en los patrones normales de desarrollo mental y social comienzan entre los seis y los 18 meses.
Cuando la mutación aparece en recién nacidos del sexo masculino la enfermedad suele ser muy agresiva, porque los niños poseen un solo Cromosoma X y éstos mueren durante los primeros
¿Qué es la enfermedad juvenil de Huntington?
La enfermedad de Huntington es un trastorno genético del cerebro que produce movimientos anormales, demencia y problemas del comportamiento. Los individuos que desarrollan la enfermedad de Huntington tienen una anormalidad en su gen de la enfermedad de Huntington desde el momento de la concepción, años o décadas antes que aparezca algún síntoma. En los Estados Unidos, la enfermedad de Huntington ocurre en 1 de cada 10,000 individuos, de casi todos los grupos étnicos. Menos de un diez por ciento de los individuos con enfermedad de Huntington presentan síntomas antes de los 20(1) años. La enfermedad juvenil de Huntington presenta desafíos únicos a los individuos afectados, a las personas que cuidan de ellos y a los diferentes profesionales que intervienen para asistirlos. Esta publicación describirá de qué manera un médico hace un diagnóstico de enfermedad Juvenil de Huntington, los síntomas que se ven con mayor frecuencia en la enfermedad juvenil de Huntington, las estrategias para afrontar la enfermedad juvenil de Huntington y el panorama actual de la investigación sobre esta enfermedad.
Cuándo se debe considerar que se trata de la enfermedad Juvenil de Huntington
No hay un síntoma ni un grupo de síntomas que sea absolutamente necesario para el diagnóstico de enfermedad juvenil de Huntington, pero hay varias características comunes en el momento del diagnóstico
Los síntomas iniciales típicos de la enfermedad
juvenil de Huntington en niños menores de 10
años son:
• Historial familiar positivo de enfermedad de
Huntington, generalmente en el padre
• Rigidez en las piernas
• Torpeza en los brazos y en las piernas
• Disminución de la habilidad mental
• Cambios en el comportamiento
• Convulsiones
• Problemas para tragar o del habla
Los movimientos como de baile o de retorcimiento (corea o baile de San Vito) que con frecuencia se ven en los adultos con enfermedad de Huntington no son comunes en los niños que desarrollan la enfermedad antes de los 10 años, pero pueden ser uno de los primeros síntomas que un adolescente exhibe. Algunas veces el primer síntoma en un adolescente son las alteraciones del comportamiento.
La enfermedad de Parkinson juvenil
Los síntomas
Los síntomas principales de Parkinson menores son, según madisonsfoundation.org, rigidez, temblores y bradykinesia de descanso. Bradykinesia es la lentitud al iniciar un movimiento.
Causas
La enfermedad de Parkinson juvenil es con frecuencia de origen desconocido (idiopática), sin embargo, hay indicios de que algunos casos son de origen genéticos. Se cree que la causa es un gen recesivo llamado Parque de 2.
Diagnóstico
Como con otras formas de Parkinson, el diagnóstico se basa en la manifestación de los síntomas. Un diagnóstico requiere dos de los tres síntomas principales (rigidez, temblores y bradykinesia de descanso) estar presente.
Tratamiento
La opción de tratamiento más frecuente para los tipos de Parkinson todos es levadopa, un medicamento que aumenta los niveles de dopamina en el cerebro. También se pueden prescribir medicamentos para ayudar a reducir los temblores (fármacos anticolinérgicos)
Prevalencia
El número exacto de las personas con Parkinson juvenil está claro, pero youngparkinsons.com informa de que la aparición del mal de Parkinson menores de 40 años se estima en aproximadamente el 10 por ciento de todos los casos.
Trastornos epilépticos
Generalidades sobre la epilepsia
La epilepsia es un trastorno cerebral en el cual una persona tiene crisis epilépticas repetidas durante un tiempo. Las crisis epilépticas son episodios de alteración de la actividad cerebral que producen cambios en la atención o el comportamiento.
Causas
La epilepsia ocurre cuando los cambios permanentes en el tejido cerebral hacen que el cerebro esté demasiado excitable o irritable. Como resultado de esto, el cerebro envía señales anormales, lo cual ocasiona convulsiones repetitivas e impredecibles. (Una sola convulsión que no sucede de nuevo no es epilepsia).
La epilepsia puede deberse a un trastorno médico o a una lesión que afecte el cerebro o la causa puede ser desconocida (idiopática).
Las causas comunes de epilepsia abarcan:
- Accidente cerebrovascular o accidente isquémico transitorio (AIT)
- Demencia, como el mal de Alzheimer
- Lesión cerebral traumática
- Infecciones, como absceso cerebral, meningitis, encefalitis y SIDA
- Problemas cerebrales presentes al nacer (anomalía cerebral congénita)
- Lesión cerebral que ocurre durante o cerca del momento del nacimiento
- Trastornos metabólicos presentes al nacer (como fenilcetonuria)
- Tumor cerebral
- Vasos sanguíneos anormales en el cerebro
- Otra enfermedad que dañe o destruya el tejido cerebral
Las crisis epilépticas por lo regular empiezan entre las edades de 5 y 20, pero pueden suceder a cualquier edad. Puede haber un antecedente familiar de convulsiones o epilepsia.
Síntomas
Los síntomas varían de una persona a otra. Algunas personas pueden tener simples episodios de ausencias, mientras otras tienen pérdida del conocimiento y temblores violentos. El tipo de convulsión o crisis epiléptica depende de la parte del cerebro afectada y la causa de la epilepsia.
La mayoría de las veces, la convulsión es similar a la anterior. Algunas personas con epilepsia tienen una sensación extraña antes de cada convulsión como hormigueo, sentir un olor que realmente no existe o cambios emocionales. Esto se denomina aura.
El médico puede darle más información acerca del tipo específico de convulsión que usted pueda tener.
- Ausencias típicas (pequeño mal) (episodios de ausencias)
- Convulsiones tonicoclónicas generalizadas (crisis de gan mal) (involucran todo el cuerpo e incluyen aura, rigidez muscular y pérdida de la lucidez mental)
- Convulsiones parciales (focales) (pueden incluir cualquiera de los síntomas anteriormente descritos, según la parte del cerebro donde se inicia la convulsión)
Pruebas y exámenes
El médico llevará a cabo un examen físico, el cual comprende una evaluación detallada del cerebro y del sistema nervioso.
Se hará un una electroencefalografía (EEG) para verificar la actividad eléctrica en el cerebro. Las personas con epilepsia generalmente tienen actividad eléctrica anormal que se observa en este examen. En algunos casos, el examen muestra el área en el cerebro donde empiezan las convulsiones. El cerebro puede aparecer normal después de una convulsión o entre convulsiones.
Para diagnosticar la epilepsia o planear la cirugía para la epilepsia, usted posiblemente necesite:
- Usar un aparato de registro electroencefalográfico durante días o semanas mientras se ocupa de su vida cotidiana.
- Permanecer en un hospital especial donde le puedan vigilar la actividad del cerebro en cámaras de video. Esto se denomina EEG en video.
Los exámenes que se pueden hacer abarcan:
- Química sanguínea
- Glucemia
- CSC (conteo sanguíneo completo)
- Pruebas de la función renal
- Pruebas de la función hepática
- Punción lumbar (punción raquídea)
- Exámenes para enfermedades infecciosas
Con frecuencia, se hace una tomografía computarizada o resonancia magnética de la cabeza para encontrar la causa y localización del problema en el cerebro.
Tratamiento
El tratamiento para la epilepsia incluye medicamentos, cambios en el estilo de vida y en ocasiones cirugía.
Si la epilepsia se debe a un tumor, vasos sanguíneos anormales o sangrado en el cerebro, la cirugía para tratar estos trastornos puede detener dichas crisis.
Los medicamentos para prevenir las convulsiones, llamados anticonvulsivos, pueden reducir el número de crisis futuras.
- Estos fármacos se toman por vía oral. El tipo de medicamento que se recete depende del tipo de convulsión que usted tenga.
- Es posible que sea necesario cambiar la dosis de vez en cuando. Usted puede necesitar exámenes de sangre para ver si hay efectos secundarios.
- Siempre tome el medicamento a tiempo y como se lo recetaron. Pasar por alto una dosis puede hacer que se presente una convulsión. Nunca deje de tomar ni cambie medicamentos por su cuenta. Hable primero con el médico.
- Muchos medicamentos para la epilepsia causan anomalías congénitas. Las mujeres que deseen quedar embarazadas deben comentarle al médico con anticipación con el fin de hacer ajustes en los medicamentos.
Muchos medicamentos para la epilepsia pueden afectar la salud de sus huesos. Hable con el médico para saber si necesita tomar vitaminas y otros suplementos.
La epilepsia que no mejora después de haber ensayado dos o tres fármacos anticonvulsivos se denomina "epilepsia resistente al tratamiento". En este caso, el médico puede recomendar una cirugía para:
- Extirpar las células cerebrales anormales que causan las convulsiones.
- Colocar un estimulador del nervio vago (ENV). Este dispositivo es similar a un marcapasos cardíaco y puede ayudar a reducir la cantidad de convulsiones.
A algunos niños se los somete a una dieta especial para ayudar a prevenir convulsiones. La más popular es la cetógena. Una dieta baja en carbohidratos, como la de Atkins, también puede servir para algunos adultos. Asegúrese de analizar estas opciones con el médico antes de ensayarlas.
Los cambios en los tratamientos médicos o en el estilo de vida pueden aumentar el riesgo de una convulsión enadultos y niños con epilepsia. Hable con su médico respecto a:
- Los nuevos medicamentos, vitaminas o suplementos recetados
- El estrés emocional
- Enfermedad, sobre todo infección
- Falta de sueño
- Embarazo
- Saltarse dosis de medicamentos para la epilepsia
- Consumo de alcohol u otras drogas psicoactivas
Trastornos tóxico-metabólico

Es un daño cerebral súbito (agudo) y problemas con la actividad hepática de causa desconocida.
El síndrome se ha presentado en niños a quienes les han administrado ácido acetilsalicílico (aspirin) cuando tenían varicela o gripe. Este síndrome se ha vuelto muy infrecuente desde que el ácido acetilsalicílico dejó de recomendarse para uso rutinario en niños.
Causas
El síndrome de Reye se observa con más frecuencia en niños de 4 a 12 años de edad. La mayoría de los casos que ocurren con varicela se dan en niños de 5 a 9 años, mientras que los que se presentan con la gripe por lo general se dan en niños de 10 a 14 años.
Síntomas
Los niños con síndrome de Reye se enferman de forma muy repentina. El síndrome a menudo comienza con vómito que dura muchas horas. El vómito es seguido rápidamente por un comportamiento irascible y agresivo. A medida que la afección empeora, el niño puede ser incapaz de permanecer despierto y alerta.
Otros síntomas del síndrome de Reye:
- Confusión
- Letargo
- Pérdida del conocimiento o coma
- Cambios en el estado mental
- Náuseas y vómitos
- Crisis epilépticas
- Posición inusual de brazos y piernas (postura de descerebración: los brazos bien extendidos apuntan hacia el cuerpo, las piernas se mantienen estiradas y los dedos de los pies apuntan hacia abajo)
Otros síntomas que pueden ocurrir con este trastorno son:
- Visión doble
- Hipoacusia
- Pérdida de la función muscular o parálisis de brazos o piernas
- Dificultades en el habla
- Debilidad en brazos o piernas
Pruebas y exámenes
Los siguientes exámenes se pueden usar para diagnosticar el síndrome de Reye:
- Análisis bioquímico de la sangre
- TC de la cabeza o IRM de la cabeza
- Biopsia del hígado
- Pruebas de la función hepática
- Examen de amoníaco sérico
- Punción raquídea
Tratamiento
No hay ningún tratamiento específico para esta afección. El médico vigilará la presión intracerebral, lagasometría arterial y el equilibrio ácido-básico (pH) de la sangre.
Los tratamientos pueden ser:
- Soporte respiratorio (se puede necesitar un respirador durante un coma profundo)
- Líquidos por vía intravenosa para suministrar electrolitos y glucosa
- Esteroides para reducir la inflamación en el cerebro
Expectativas (pronóstico)
El pronóstico de una persona depende de la severidad del coma al igual que de otros factores.
El pronóstico para aquellos que sobreviven a un episodio agudo puede ser bueno.
Posibles complicaciones
- Coma
- Daño cerebral permanente
- Crisis epilépticas
Sin tratamiento, las crisis epilépticas y el coma pueden ser potencialmente mortales
Los errores congénitos del metabolismo

{kind=link}
(ECM) pueden debutar en la adolescencia y en la edad adulta. Aunque es difícil aportar datos exactos de prevalencia ya que existen escasos estudios al respecto, incluso considerándolos poco frecuentes, la importancia de su detección radica en las posibilidades terapéuticas y de consejo genético familiar.
La principal sintomatología de los ECM del adulto es la neurológica, seguida de la hepática. Se puede establecer dos modos básicos de debut. Uno es el agudo, normalmente en forma de alteración del nivel de conciencia, letargia, coma de etiología desconocida en un paciente previamente sano (déficits del ciclo de la urea, trastornos de la remetilación de la homocisteína y porfirias son aquí las causas más frecuentes). Por otra parte está la sintomatología crónica, insidiosa, a menudo progresiva, en la que suele haber cuadros clínicos complejos, y más raramente un síntoma aislado de manera persistente (la enfermedad de Wilson, enfermedades mitocondriales, lisosomales, la enfermedad de Refsum y las glucogenosis son algunos ejemplos en este grupo).
Es de especial importancia conocer las formas de debut agudo, que suelen ser situaciones de extrema urgencia, en las que una conducta adecuada puede evitar el fallecimiento del paciente. En este caso, exámenes sencillos de laboratorio como la determinación del amonio, homocisteína, lactato, acilcarnitinas, aminoácidos, ácidos orgánicos y porfirinas, pueden orientar el diagnóstico y permiten iniciar un tratamiento intensivo.
En este capítulo se pretende realizar un enfoque práctico, abordando las características generales y claves clínicas de sospecha de los ECM más habituales en el adulto.
La principal sintomatología de los ECM del adulto es la neurológica, seguida de la hepática. Se puede establecer dos modos básicos de debut. Uno es el agudo, normalmente en forma de alteración del nivel de conciencia, letargia, coma de etiología desconocida en un paciente previamente sano (déficits del ciclo de la urea, trastornos de la remetilación de la homocisteína y porfirias son aquí las causas más frecuentes). Por otra parte está la sintomatología crónica, insidiosa, a menudo progresiva, en la que suele haber cuadros clínicos complejos, y más raramente un síntoma aislado de manera persistente (la enfermedad de Wilson, enfermedades mitocondriales, lisosomales, la enfermedad de Refsum y las glucogenosis son algunos ejemplos en este grupo).
Es de especial importancia conocer las formas de debut agudo, que suelen ser situaciones de extrema urgencia, en las que una conducta adecuada puede evitar el fallecimiento del paciente. En este caso, exámenes sencillos de laboratorio como la determinación del amonio, homocisteína, lactato, acilcarnitinas, aminoácidos, ácidos orgánicos y porfirinas, pueden orientar el diagnóstico y permiten iniciar un tratamiento intensivo.
En este capítulo se pretende realizar un enfoque práctico, abordando las características generales y claves clínicas de sospecha de los ECM más habituales en el adulto.
ECM en el niño y en el adulto: rasgos diferenciales básicos
Los trastornos del metabolismo de las moléculas complejas (lisosomales, peroxisomales) y los déficits del metabolismo energético (enfermedades mitocondriales) son los principales grupos de patologías que se presentan en la edad adulta. Cabe destacar además otros trastornos que de manera aislada también se presentan con frecuencia en esta etapa de la vida como la enfermedad de Wilson (metabolismo de los metales) y la enfermedad de Segawa o déficit de GTPCH1 de transmisión dominante (metabolismo de los neurotransmisores).
En el adulto, los ECM suelen aparecer en forma de patologías insidiosas, lentamente progresivas y con mucha menor frecuencia en forma de descompensación aguda o desencadenados por algún agente externo. No obstante, situaciones especialmente catabólicas como el parto, intervenciones quirúrgicas, ejercicio físico intenso o infecciones sistémicas pueden ser los detonantes de estos déficits. Además, el mismo tipo de error congénito puede manifestarse con una clínica totalmente diferente en el adulto que en el niño2. La tabla 1 refleja las enfermedades más prevalentes en función del grupo bioquímico al que corresponden, así como las principales diferencias de presentación en función de la edad.
Mal nutrición

Condición física caracterizada por un desorden nutricional, el cual es producido por una alimentación insuficiente o inadecuada que no aporta todos los nutrientes necesarios para una vida activa y saludable.
Aunque frecuentemente los conceptos de malnutrición y desnutrición se utilizan indistintamente, el primero es más amplio. En efecto, la malnutrición es un desorden nutricional que, según Foster (1992:13-29), puede ser de diferentes tipos:
a) Sobrealimentación: por exceso de consumo de calorías, frecuente en los países desarrollados.
b) Desnutrición: producida por un consumo insuficiente de calorías y proteínas para garantizar las funciones del cuerpo, su crecimiento y una actividad física normal.
c) Deficiencia dietética: falta en la dieta de determinados micronutrientes esenciales, como minerales y vitaminas.
d) Malnutrición secundaria: causada no por la dieta, sino por enfermedades o patologías que impiden al organismo absorber los nutrientes ingeridos (diarrea, infecciones, sarampión, parásitos intestinales, etc.), lo cual contribuye a la desnutrición.
Las tres últimas son habituales en los países pobres, siendo la más relevante la desnutrición, también llamada Malnutrición Proteico-Energética (MPE). Esas tres situaciones, y principalmente la desnutrición, suelen denominarse coloquialmente como hambre. Cuando en un lugar y momento dados ésta experimenta un proceso de agravamiento que se ve acompañado de otros factores (empobrecimiento, epidemias, frecuentemente aumento de mortalidad), nos encontramos ante una hambruna.
Según la FAO (1999:4,11), unos 790 millones de personas padecían malnutrición en los países en desarrollo en 1995-97, a los que se añaden otros 34 millones en los desarrollados, sobre todo en los países en transición del Centro y Este de Europa. Según unicef (1998:16-17), un total de 226 millones de niños menores de cinco años (el 40% de este grupo) sufren desnutrición, lo que les acarrea un retraso entre moderado y grave en su crecimiento. De ellos, algunos viven en los países ricos, por ejemplo 13 millones en EE.UU. (UNICEF, 1998:16-17).
Tanto la incidencia actual del problema como su evolución reciente y las perspectivas futuras varían ampliamente de unos continentes a otros: Es en Asia donde habitan más personas malnutridas pero en el que se han registrado mayores avances en las últimas tres décadas, mientras que en África los malnutridos representan un mayor porcentaje de la población, habiendo tenido una evolución reciente negativa y siendo la única región para la que se espera un empeoramiento en los próximos años (respecto a la magnitud y causas del problema, ver hambre).
La malnutrición afecta a todos los grupos de edad, pero algunos son particularmente vulnerables, sobre todo los niños (en especial entre los 6 meses y cinco años, después del destete), las mujeres embarazadas y las que amamantan. A éstos se añaden los ancianos y los discapacitados físicos.
1) Efectos de la Malnutrición Proteico-Energética
La MPE, o insuficiencia de calorías y proteínas en la dieta, da lugar a diferentes problemas, entre los que destacan los siguientes:
a) Deterioro de la salud, que a su vez daña el estado nutricional: la desnutrición disminuye la capacidad inmunológica del organismo, facilitando las infecciones, que provocan malnutrición secundaria (merma de la absorción de nutrientes), dando lugar a un círculo vicioso.
b) Aumento de la mortalidad, sobre todo entre los niños: generalmente no por la desnutrición en sí, sino por las enfermedades derivadas de la debilidad inmunológica que aquélla provoca. Destacan sobre todo las infecciones diarreicas, así como también la neumonía, la gripe, la bronquitis o el sarampión. Todas estas enfermedades, fácilmente evitables, representan más del 40% de la mortalidad infantil en el tercer mundo. Además, la malnutrión en mujeres embarazadas, aparte de afectar al desarrollo del feto, incrementa su riesgo de enfermedad y muerte por causas relacionadas con el embarazo y el parto.
c) Retrasos en el crecimiento de los niños, sobre todo por el insuficiente consumo de calorías, que pueden manifestarse en dos formas, o en la combinación de ambas:
– El retraso en el crecimiento o desmedro __: significa que el niño tiene una estatura menor a la que corresponde para su edad, e indica que su alimentación ha sido inadecuada durante un largo período, aunque en la actualidad pueden ser niños completamente sanos. Ese retraso en el crecimiento puede no ser reversible, de forma que su altura final en la adultez será finalmente menor de lo que debiera.
– La _emaciación _: significa que el peso del niño es inferior al que corresponde para su altura, lo que implica que en ese momento está o que recientemente ha estado sufriendo de inanición o de enfermedad. La emaciación toma frecuentemente un patrón estacionario (en los meses previos a la cosecha, de cierta escasez alimentaria), especialmente en las economías de subsistencia; y aunque es un signo de alerta, puede ser fácilmente reversible una vez que el niño tiene lo suficiente para comer y no tiene ninguna enfermedad que le impida alimentarse y/o asimilar los alimentos.
La __malnutrición proteico-energética moderada está bastante extendida y afecta a una buena parte de la población de los países pobres, pasando muchas veces desapercibida a no ser que se realicen mediciones regulares, sobre todo en los niños. Para ello se utilizan diferentes mediciones antropométricas, como el bajo peso al nacer, la baja altura por la edad, el bajo peso por la altura, o el perímetro del brazo en su punto medio, mediciones que son utilizadas como parte de los sistemas de alerta temprana de seguridad alimentaria. Aunque no sea grave, puede contribuir a la mortalidad en la medida en que debilita la resistencia ante las infecciones.
La malnutrición proteico-energética grave, por el contrario, ocasiona síntomas clínicos fácilmente observables, al tiempo que constituye, junto a las epidemias, una de las principales causas del incremento de la mortalidad en la fase de emergencia durante un proceso de hambruna. Tal malnutrición grave frecuentemente da lugar a dos cuadros clínicos bien definidos, el marasmo y el kwashiorkor, o a variaciones o combinaciones de ambos.
Deprivación ambiental
Desventaja psicosocial
El período preescolar es una etapa de adquisiciones en las esferas del desarrollo físico y psíquico, de ellos se desprende que necesitan una atención especial para tratar de compensar sus deficiencias físicas y emocionales, atención que debe estar inmersa de un profundo afecto y dedicación por parte de la familia, por ello las relaciones padre e hijo deben ser portadoras de un fuerte apoyo, de una manifestación incondicional que el niño se conozca a sí mismo y vaya adquiriendo seguridad de sus propias fuerzas.
Cuando esto ocurre, el niño es capaz, en el seno de su familia, de relatar las experiencias adquiridas durante el día en el centro infantil; y establece así nuevas relaciones ampliando su conocimiento del mundo.
La influencia familiar es determinante en el desarrollo del individuo y en particular en las primeras edades, donde se forman las premisas del desarrollo de la personalidad y se inicia la formación de sus cualidades psíquicas; sin embargo, en ocasiones nos encontramos que existen familias en las que esta influencia es adversa, dado que no les garantizan al niño las condiciones de vida, alimentación, afecto y cuidado, y en otras puede tener un carácter deficitario, por no estar totalmente satisfechas estas necesidades básicas. En uno u otro caso existe un común denominador y es la desatención de padres a hijos.
Cuando el niño se mantiene en un medio de hipoestimulación en el que no se le satisface la necesidad de comunicación de estimulación y afecto , que resulta vital desde las primeras edades, se observa rápidamente una repercusión negativa que de mantenerse podría ocasionar daños irreparables a la salud del niño.
La falta de estimulación generalmente se acompaña de una déficit en la relación afectiva. El niño necesita sentirse querido y protegido por los padres o adultos que cuidan de él. Cuando en esta relación se ponen de manifiesto actitudes de rechazo, descuido, negligencia, pobreza del medio, tanto en cuanto a estimulación como a posibilidades de actividades, el desarrollo físico y psíquico del niño se ve afectado.
El adulto es el encargado de organizar la vida de los niños, es por esta vía que ellos se van relacionando con el medio que los rodea, con sus objetos y múltiples relaciones; medio que significa una fuente de estímulos que le llega al niño, en un inicio mediatizados por el adulto. Por eso afirmamos que es en la relación adulto-niño, en esa directa y estrecha comunicación, que debe producirse la asimilación por el niño de la experiencia histórica social de la humanidad.
Análisis particular requiere las graves consecuencias derivadas de condiciones desfavorables por carencia de estímulos, afecto y comunicación cuando estas se producen en las primeras etapas de vida. Por ejemplo, el primer año de vida se caracteriza por un ritmo veloz de crecimiento y desarrollo, por una estrecha relación entre el desarrollo neuropsíquico y físico y porque los niños presentan poca resistencia ante las enfermedades; dadas estas características, la educación en esta edad debe contemplar el cumplimiento riguroso del horario de vida, propiciar las condiciones que favorecen el desarrollo óptimo del niño en la edad. Cuando uno de los aspectos señalados anteriormente es insuficiente o está ausente se afecta el desarrollo, bien porque lo frena o lo limita.
Cuando los niños están desprovistos de la necesaria estimulación desde las edades tempranas, o esta ha sido insuficiente, se aprecia una repercusión desfavorable en su desarrollo, y si esto se conjuga con una pobre relación afectiva se presentan consecuencias más severas, tales como retardo de los movimientos y del desarrollo del lenguaje, de las posibilidades de aprendizaje, de la expresión afectiva, pudiendo llegar a situaciones severas de retardo general del desarrollo.
Entre los estudios realizados sobre efectos de condiciones deficitarias de vida se encuentran las efectuadas por R. Spitz (1). En sus trabajos encontramos un estudio de las consecuencias de las enfermedades defectivas emocionales en niños del primer año de vida sometidos a condiciones de privación total o parcial de afecto y estimulación.
Al estudiar la etiología de las enfermedades defectivas emocionales. Spitz afirma que estas se derivan, por lo general, de la ausencia física materna y que el sustituto de la madre es inadecuado o prácticamente no existe. Considera como aspecto esencial “la no relación” con la madre y le otorga un “papel secundario” a la personalidad individual de la misma. El daño sufrido por el niño privado de su madre será proporcional al período en que transcurre esta privación y distingue las categorías o momentos de las enfermedades defectivas emocionales: la privación afectiva o depresión analítica y la privación afectiva total que la denomina hospitalismo o institucionalismo. Señala que no existe una división entre estos dos síndromes resultantes de la privación afectiva y que se da una transición de uno a otro, con un carácter progresivo, atravesando fases de severidad crecientes. Los síntomas se van haciendo más agudos en dependencia del aumento del período de separación, llegando hasta el hospitalismo, fase más aguda en la que se produce un empeoramiento progresivo de la salud del niño y aumenta la propensión a las infecciones, que lo puede llevar al marasmo y a la muerte.
En los trabajos de R. Spitz se centra la atención en el aspecto biológico de la relación "madre-hijo", sin destacar el contenido social de esta relación. Los niños estudiados habían sido separados de sus madres e internados en centros con características hospitalarias, se constató que ellos mismos empeoraban en la medida que aumentaba el tiempo de estancia; y que después de un período de separación de cinco meses el proceso de la enfermedad se hacía irreversible.
En nuestro país durante la etapa de la república neocolonial existió la antigua Casa de Beneficiencia (asilo de expósitos), en esta como en otros "asilos" infantiles los niños se encontraban en condiciones de hipoestimulación, vivían en pabellones de los cuales prácticamente no salían, carecían de juguetes y eran cuidados por niñeras poco interesadas en ellos. Los resultados de estudios realizados en Cuba por J. Pérez Villar mostraron que todos los niños estudiados presentaban graves trastornos de la personalidad, y la gran mayoría un considerable déficit intelectual (2). Las pésimas condiciones de vida, afecto o estimulación afectaban sensiblemente el desarrollo de los niños y la formación de cualidades de la personalidad.
Muchos de aquellos niños que se criaron en orfanatos y asilos para huérfanos presentaban conductas similares de las descritas por Spitz. En estas instituciones el pronóstico del desarrollo de los niños era desfavorable, dado por la ausencia de la relación afectiva directa con la madre y por las condiciones en que transcurría su vida, donde no existía un trabajo educativo ni pedagógico dirigido a suplir y contrarrestar la ausencia o insuficiencia del cuidado materno, por lo que no era extraño encontrar niños con verdaderas manifestaciones de institucionalismo.
Con el triunfo de la Revolución en Cuba en 1959 se producen grandes transformaciones de carácter social y entran en vigor programas de desarrollo educacional. Quedan atrás los asilos, las casas cunas y los hogares infantiles que eran verdaderos almacenes de niños. Se inicia una etapa de intenso trabajo dirigido a la transformación de estos tipos de instituciones y en el que la Federación de Mujeres Cubanas jugó un papel decisivo, tanto en la creación de nuevas condiciones materiales y de estimulación, como en un adecuado clima afectivo que garantizara la atención educativa. Se realizó un enorme esfuerzo en la selección y preparación del personal que atendiera y cuidara a niños sin amparo filial en centros preescolares concebidos con un carácter formativo y educativo, y de desarrollo de las amplias potencialidades de cada niño en particular. Surgen los círculos infantiles en 1961. (3)
Paralelamente al desarrollo de los círculos infantiles se desplegó una amplia labor encaminada a la educación de padres, con el objetivo de que estos participaran activamente en el trabajo organizativo y educacional de las instituciones preescolares a los que asistían sus hijos. Esta labor estuvo dirigida desde sus inicios a lograr una sólida unión entre la educación familiar y social.
Se considera de gran importancia la relación del niño con su medio familiar, y muy especialmente con la figura de la madre. En la atención al niño el cuidado afectuoso y estimulante debe ir acompañado de condiciones de vida y educación favorables, ya que estas son determinantes para un sano desarrollo de la personalidad del pequeño
castigos y falta de cuidados en la infancia, deficiencias sociales, sensoriales crónicas

.
TIPOS DE MALTRATO
Existen diferentes tipos de maltrato, definidos de múltiples formas, nosotros hemos seleccionado los siguientes:
Maltrato físico
Se define como maltrato físico a cualquier lesión física infringida al niño/a
(hematomas, quemaduras, fracturas, u otras lesiones) mediante pinchazos, mordeduras, golpes, tirones de pelo, torceduras, quemaduras, puntapiés u otros medios con que se lastime el niño.
En la definición del maltrato infantil es necesario recalcar el carácter intencional, nunca accidental, del daño o de los actos de omisión lle adas a cabo por los responsables del cuidado del niño/a, con el propósito de lastimarlo o injuriarlo.
Aunque el padre o adulto a cargo puede no tener la intención de lastimar al niño, también se interpreta como maltrato a la aparición decualquier lesión física arriba señalada que se produzca por el empleo de algún tipo de castigo inapropiado para la edad del niño/a.
A diferencia del maltrato físico el castigo físico se define como el empleo de la fuerza física con intención de causar dolor, sin lesionar, con el propósito de corregir o controlar una conducta. No siempre es sencillo saber cuando termina el "disciplinamiento" y comienza el abuso. En contraposición con el maltrato físico, el castigo corporal es una práctica muy difundida y socialmente aceptada. A pesar de ello, constituye una iolación de los derechos fundamentales como personas, es un atentado contra su dignidad y autoestima, es una práctica peligrosa porque puede causar daños gra es a los niños y constituye siempre una forma de abuso psicológico que puede generar estrés y depresiones. Los niños que sufren este tipo de castigo tienden a reproducir comportamientos antisociales y a con ertirse en adultos iolentos.
Las estadísticas acerca del maltrato físico de los niños son alarmantes. Se estima que cientos de miles de niños han recibido abuso y maltrato a manos de sus padres o parientes. Miles mueren. Los que sobre i en el abuso, i en marcados por el trauma emocional, que perdura mucho después de que los moretones físicos hayan desaparecido. Las comunidades y las cortes de justicia reconocen que estas Adheridas emocionales ocultas pueden ser tratadas. El reconocer y dar tratamiento inmediato es importante para minimizar los efectos a largo plazo causados por el abuso o maltrato físico.
SIGNOS DE ABUSO FÍSICO
Quemaduras, mordeduras, fracturas, ojos morados, o dolores en el niño que aparecen bruscamente y no tienen una explicación con incente.
Hematomas u otras marcas e identes luego de haber faltado a clases.
Parece temerle a sus padres y protesta o llora cuando es hora de dejar el colegio para ir a su casa.
Le teme al acercamiento o contacto de otros mayores.
Nos dice que le han pegado en su casa.
Ante una lesión o traumatismo e idente en el niño/a no brindan una explicación con incente o se enojan ante la pregunta de lo ocurrido.
Frecuentemente se refieren a su hijo/a como "un demonio" o en alguna otra manera despecti a.
Es frecuente er que tratan al niño/a con disciplina física muy dura.
Sus padres tienen antecedentes de haber sido niños maltratados o abandonados.
Existe el antecedente de que la madre a sido golpeada.
Es frecuente que la madre del niño concurra con algún moretón u "ojo en compota".
ABANDONO O NEGLIGENCIA
Significa una falla intencional del los padres o tutores en satisfacer las necesidades básicas del niño en cuanto a alimento, abrigo o en actuar debidamente para sal aguardar la salud, seguridad, educación y bienestar del niño.
Pueden definirse dos tipos de abandono o negligencia:
Abandono Físico: Este incluye el rehuir o dilatar la atención de problemas de salud; echar de casa a un menor de edad; no realizar la denuncia o no procurar el regreso al hogar del niño/a que huyó; dejar al niño solo en la casa o a cargo de otros menores.
Negligencia o Abandono Educacional: No inscribir a su hijo en los ni eles de educación obligatorios para cada pro incia; no hacer lonecesario para pro eer la atención a las necesidades de educación especial.
En di ersas oportunidades realizar el diagnóstico de negligencia o descuido puede presentar problemas de subjeti idades. El descuido puede ser intencional como cuando se deja a un niño sin comer como castigo, o no intencional como cuando se deja solo a un niño durante horasporque ambos padres trabajan fuera del hogar. En este último ejemplo como tantos otros que genera la pobreza, el abandono o descuido es más un resultado de naturaleza social que de maltrato dentro de la familia.
Signos de Negligencia
Falta frecuentemente a la escuela.
Pide o roba plata u otros objetos a compañeros de colegio.
Tiene serios problemas dentales o isuales y no recibe tratamiento acorde.
Es habitual que concurra a clases sucio, o con ropa inadecuada para la estación, sin que la condición de sus padres sea la de pobrezaextrema.
Hay antecedentes de alcoholismo o consumo de drogas en el niño o la familia.
El niño/a comenta que frecuentemente se queda solo en casa o al cuidado de otro menor.
Se muestran indiferentes a lo que los docentes dicen del niño/a.
Su comportamiento en relación al niño/a o la institución es irracional.
Padecen de alcoholismo u otra dependencia.
Tienen una situación socio económica que no explica el descuido en la higiene, el uso de estimenta inadecuada para la estación, o la imposibilidad de solución de algunos de sus problemas de salud que presenta el niño/a.
Abuso Sexual
Puede definirse como tal a los contactos o acciones recíprocas entre un niño/a y un adulto, en los que el niño/a está siendo usado para gratificación sexual del adulto y frente a las cuales no puede dar un consentimiento informado. Puede incluir desde la exposición de los genitales por parte del adulto hasta la iolación del niño/a. La mayoría de estos delitos se producen en el ámbito del hogar, siendo el abusadormuchas eces un miembro de la familia o un conocido de esta o el menor.
Una forma común de abuso sexual es el incesto, definido este como el acto sexual entre familiares de sangre, padre hija, madre hijo, entre hermanos.
Los niños que han sido abusados pueden exhibir:
Una pobre auto imagen
Reactuación del acto sexual
Conducta agresi a, problemas de disciplina y, a eces, comportamiento ilegal
Coraje y rabia
Comportamiento auto destructi o o auto abusi o, pensamientos suicidas
Pasi idad y comportamiento retraído
Miedo de establecer relaciones nue as o de comenzar acti idades nue as
Ansiedad y miedos
isiones de experiencias ya i idas y pesadillas
Abuso de drogas o de alcohol
A menudo el daño emocional se ero a los niños maltratados no se refleja hasta la adolescencia, o aún más tarde, cuando muchos de estos niños maltratados se con ierten en padres abusi os y comienzan a maltratar a sus propios hijos. Un adulto que fue abusado de niño tiene mucha dificultad para establecer relaciones personales íntimas. Estas íctimas, tanto hombres como mujeres, pueden tener problemas para establecer relaciones cercanas, para establecer intimidad y confiar en otros al llegar a adultos; están expuestos a un riesgo mayor de ansiedad.
Signos de Abuso Sexual
Es necesario remarcar que el grado de afectación o impacto sobre la niña/o depende de arios factores como quien perpetró el abuso, la cronicidad del hecho, la utilización de fuerza, la personalidad particular de la niña/o abusada/o, su edad o sexo, etc. Es por ello que la niña/o abusada/o puede responder de ariadas formas.
Tiene dificultades para sentarse o caminar.
Repentinamente no quiere hacer ejercicios físicos.
Demuestra comportamientos o conocimientos sexuales inusuales o sofisticados para la edad.
Tiene o simula tener acti idad sexual con otros compañeros menores o de la misma edad.
Queda embarazada o contrae enfermedades de transmisión sexual antes de los 14 años.
Hay antecedentes de haber huido de la casa.
Son extremadamente protectores del niño/a
Ninguno de estos signos por si solo demuestra o prueba que el maltrato físico esta presente en la casa de este niño. Alguno de estos signos suelen hallarse en algún momento, en algún niño o familiar. Cuando los mismos aparecen en forma repetida o se combinan entre sí, esnecesario que el docente considere la posibilidad que este niño este sufriendo algún tipo de maltrato e intente algún acercamiento más intimocon él y su situación.
Maltrato Emocional
Esta es una de las formas más sutiles pero también más extendidas de maltrato infantil. Son niños/as habitualmente ridiculizados, insultados, regañados o menospreciados. Se los somete en forma permanente a presenciar actos de iolencia física o erbal hacia otros miembros de la familia. Se les permite o tolera el uso de drogas o el abuso de alcohol. "Si bien la ley no define el maltrato psíquico, se entiende como tal a toda aquella acción que produce un daño mental o emocional en el niño, causándole perturbaciones de magnitud suficiente paraafectar la dignidad, alterar su bienestar o incluso perjudicar su salud".
Actos de pri ación de la libertad como encerrar a un hijo o atarlo a una cama, no solo pueden generar daño físico, sino seguramente afecciones psicológicas se eras. Lo mismo ocurre cuando se amenaza o intimida permanentemente al niño, alterando su salud psíquica. Estos dos últimos ejemplos están contemplados como iolación al código penal.
Signos de Maltrato Emocional
Considere la posibilidad de maltrato emocional cuando el niño/a.
Muestra comportamientos extremos, algunas eces una conducta que requiere llamados de atención y otras pasi idad extrema.
Asume tanto roles o actitudes de "adulto", como por ejemplo cuidar de otros niños, como otras demasiado infantiles para su edad.
Muestra un desarrollo físico o emocional retrasado.
Ha tenido intentos de suicidio.
Considere la posibilidad de maltrato emocional cuando sus padres o tutores:
Constantemente menosprecian, o culpan al niño/a.
No les importa lo que pasa o les dicen los maestros acerca del niño, o se niegan a considerar la ayuda que le ofrecen para superar los problemas del niño en el colegio.
Abiertamente rechazan al niño/a.
Otros Tipos de Maltratos
Ø Abandono emocional: Situación en la que el niño no recibe el afecto, la estimulación, el apoyo y protección necesarios en cada estadio de su e olución y que inhibe su desarrollo óptimo. Existe una falta de respuesta por parte de los padres/madres o cuidadores a las expresiones emocionales del niño (llanto, sonrisa,...) o a sus intentos de aproximación o interacción.
Ø Síndrome de M nchhausen por poderes: Los padres/madres cuidadores someten al niño a continuas exploraciones médicas, suministro de medicamentos o ingresos hospitalarios, alegando síntomas ficticios o generados de manera acti a por el adulto (por ejemplo mediante laadministración de sustancias al niño).
Ø Maltrato institucional: Se entiende por malos tratos institucionales cualquier legislación, procedimiento, actuación u omisión procedente de los poderes públicos o bien deri ada de la actuación indi idual del profesional que comporte abuso, negligencia, detrimento de la salud, la seguridad, el estado emocional, el bienestar físico, la correcta maduración o que iole los derechos básicos del niño y/o la infancia.
Signos para sospechar maltrato infantil
En el niño/a:
Muestra repentinos cambios en el comportamiento o en su rendimiento habitual.
Presenta problemas físicos o médicos que no reciben atención de sus padres.
Muestra problemas de aprendizaje que no pueden atribuirse a causas físicas o neurológicas.
Siempre esta "expectante", como preparado para que algo malo ocurra.
Se e idencia que falta super isión de los adultos.
Es sumamente hiperacti o o por el contrario excesi amente responsable.
Falta en forma reiterada al colegio.
En la familia:
Dan muestras de no preocuparse por el hijo; raramente responden a los llamados del colegio o al cuaderno de citaciones.
Niegan que el niño tenga problemas, tanto en el colegio como en el hogar, o por el contrario maldicen al niño por su conducta.
Por su propia oluntad autorizan a que la maestra emplee "mano dura" o incluso algún chirlo o sacudón si su hijo se porta mal.
Cuestionan todo lo que hace su hijo, se burlan o hablan mal de él ante los maestros.
Demandan de su hijo un ni el de perfección académica o un rendimiento físico que es inalcanzable para el niño.
En los padres e hijos:
Consideran que la relación con su hijo es totalmente negati a.
Demuestran que casi nadie les cae bien.
Tienen una actitud recíproca de permanente tensión.
Indicadores de Maltrato Infantil
El niño no sabe defenderse ante las agresiones de los adultos, no pide ayuda, esto lo sitúa en una posición ulnerable ante un adulto agresi o y/o negligente. Los niños que sufren maltrato tienen múltiples problemas en su desarrollo e oluti o, déficit emocionales, conductuales y socio cogniti os que le imposibilitan un desarrollo adecuado de su personalidad. De ahí la importancia de detectar cuanto antes el maltrato y buscaruna respuesta adecuada que ayude al niño en su desarrollo e oluti o.
Los problemas que tienen los niños maltratados se traducen en unas manifestaciones que pueden ser conductuales, físicas y/o emocionales. A estas señales de alarma o pilotos de atención es a lo que llamamos indicadores, ya que nos pueden "indicar" una situación deriesgo o maltrato.
A continuación exponemos una serie de indicadores que nos pueden ayudar en nuestra obser ación, sin embargo hay que tener en cuentaque éstos por sí solos no son suficientes para demostrar la existencia de maltrato sino que además debemos considerar la frecuencia de las manifestaciones, cómo, dónde y con quién se producen.
Por ello es importante saber interpretar estos indicadores y no quedarnos ante ellos como obser adores o jueces de una forma de ser ante la que no podemos hacer nada. Estos indicadores no siempre presentan e idencias físicas ( algunas formas de abuso sexual, maltrato psicológico...) sino que pueden ser también conductas difíciles de interpretar.
Algunos de los indicadores, entre otros, que se pueden dar son:
En el Niño:
Señales físicas repetidas (morados, magulladuras, quemaduras...)
Niños que an sucios, malolientes, con ropa inadecuada, etc.
Cansancio o apatía permanente (se suele dormir en el aula)
Conductas agresi as y/o rabietas se eras y persistentes
Relaciones hostiles y distantes
Actitud hiper igilante (en estado de alerta, receloso,...)
Conducta sexual explícita, juego y conocimientos inapropiados para su edad
Conducta de masturbación en público
Tiene pocos amigos en la escuela
Muestra poco interés y moti ación por las tareas escolares
Después del fin de semana uel e peor al colegio (triste, sucio, entre otro)
Presenta dolores frecuentes sin causa aparente
Falta a clase de forma reiterada sin justificación
Retrasos en el desarrollo físico, emocional e intelectual
Presenta conductas antisociales: fugas, andalismo, pequeños hurtos, entre otros.
Intento de suicidio y sintomatología depresi a
Regresiones conductuales (conductas muy infantiles para su edad)
Relaciones entre niño y adulto secreta, reser ada y excluyente
Falta de cuidados médicos básicos
En los Padres y/o Cuidadores:
Parecen no preocuparse por el niño
No acuden nunca a las citas y reuniones del colegio
Desprecian y des alorizan al niño en público
Sienten a su hijo como una "propiedad" ("puedo hacer con mi hijo lo que quiero porque es mío")
Expresan dificultades en su matrimonio
Abusan de substancias tóxicas (alcohol y/o drogas)
Trato desigual entre los hermanos
No justifican las ausencias de clase de sus hijos
Justifican la disciplina rígida y autoritaria
en al niño como mal ado
Ofrecen explicaciones ilógicas, contradictorias no con incentes o bien no tienen explicación
Habitualmente utilizan una disciplina inapropiada para la edad del niño
Son celosos y protegen desmesuradamente al niño
Síndrome de hipoconexión.
Cuando existe clara presunción de retraso mental, pero la inteligencia del sujeto no puede ser evaluada mediante los test. Usuales (individuos no cooperadores).
*Retraso Mental Leve.
- Se considera en la categoría pedagógica como educables.
- Incluye la mayoría (el 85 por 100). De las personas afectadas.
- Durante su vida adulta acostumbran a adquirir habilidades sociales y laborables adecuadas para una autonomía mínimo pero pueden necesitar supervisión, y asistencia en situaciones de estrés social o económico desusado.
*Retraso Mental Moderado.
- Este grupo constituye alrededor del 10 por 100 de toda la población afectada.
- La mayoría de los individuos con este nivel de retraso mental adquieren habilidades de comunicación durante los primeros años de la niñez.
- Pueden aprovecharse de una forma laboral, y con supervisión moderada, atender a su propio cuidado personal.
*Retraso Mental Grave.
- Incluyen el 3-4 por 100 de los individuos con Retraso Mental.
- Durante los primeros años de la niñez adquieren un lenguaje comunicativo escaso o nulo.
- En su mayoría se adaptan bien a la vida en la comunidad, sea en hogares colectivos o con sus familias, a no ser que sufran alguna discapacidad asociada que requiera cuidados especializados o cualquier tipo de asistencia.
*Retraso Mental Profundo.
- Incluye aproximadamente, el 1-2 por 100 de las personas con retraso mental.
- La mayoría presenta una enfermedad neurológica identificada que explica su retraso mental.
- Durante los primeros años de la niñez desarrollan considerables alteraciones del funcionamiento sensorio-motor.
- Llegan a realizar tareas simples y estrechamente supervisados.